27, Feb 2026
De validar métodos analíticos con hojas de cálculo, a tener el control total.
Validar métodos analíticos utilizando hojas de cálculo no validadas (como archivos de Excel sin control) representa uno de los mayores riesgos de integridad de datos en laboratorios regulados (GMP/GLP/ISO 17025). Los principales problemas se centran en la falta de fiabilidad, la vulnerabilidad de los datos y el incumplimiento normativo. En los procesos de validación de métodos analíticos, no solo es importante la obtención de un reporte con buenos resultados. La pregunta es; si los datos obtenidos aún asi sea los esperados o no, estos son procedentes de un lugar seguro, integro y controlado. Para lograr establecer el dónde se está posicionado, es necesario conocer los riesgos principales en integridad de datos que se pueden presentar en nuestras hojas de cálculo. A continuación, se detallan los riesgos principales existentes en hojas de cálculo:
-Falta de integridad de los datos y auditoría: Las hojas no validadas suelen carecer de audit trails (pistas de auditoría) automáticas, lo que impide rastrear quién realizó cambios, cuándo o por qué. Esto contraviene los principios ALCOA+ (Atribuible, Legible, Contemporáneo, Original, Exacto), establecidos por las regulaciones CFR21 parte 11.
-Errores ocultos en fórmulas y cálculos: Sin validación, los errores en las celdas (fórmulas incorrectas, referencias cruzadas equivocadas, errores de redondeo, condicionales mal argumentadas) pueden pasar desapercibidos, resultando en cálculos de concentración, exactitud o precisión incorrectos.
-Modificación no autorizada de datos: Las hojas de cálculo no protegidas permiten que las fórmulas o celdas críticas sean editadas por error o malintencionadamente, cambiando los resultados finales sin dejar rastro.
-Falta de control de versiones: El uso de archivos con nombres de identificación poco claros como, por ejemplo; «metodo_final_v2_final.xlsx» lleva a la confusión sobre cuál es la plantilla oficial, provocando que se validen métodos con versiones obsoletas.
-Seguridad y accesibilidad inadecuadas: Los archivos suelen guardarse en computadoras locales sin respaldo (back-up) ni control de acceso, lo que pone en riesgo la pérdida de información o la manipulación de datos.
-Consecuencias regulatorias: Las agencias (FDA, EMA) sancionan frecuentemente a los laboratorios que usan Excel para cálculos críticos sin una validación formal, lo que puede resultar en la invalidación de reportes de control de calidad o en su caso el levantamiento de una carta de advertencia 483, el cual es un documento oficial que levanta la FDA detallando las deficiencias graves encontradas durante una inspección en instalaciones reguladas.
Tal y como se describió, el uso de hojas de cálculo debe ser menos mecánico y ser más consciente del impacto en la seguridad de los datos que estos archivos pueden presentar.
Lleva el Control de validaciones de métodos analíticos
En Deappharma desarrollamos eADMXL, un sistema especializado apegado a las regulaciones internacionales para elevar el control de datos y registros de validación de métodos analíticos. El control se lleva mediante la estructuración de registros y módulos de trabajo, tales cuales, permiten la captura de información asociada al proyecto, data cruda y documentos. La estructuración del sistema, eADMxL, mediante módulos lógicos limita el acceso a fórmulas y solo permite la obtención de resultados en formato PDF (protegidos). Para con esto, mitigar los eventos asociados a la integridad de datos por las actuales rutinas de trabajo. eADMxL, es un sistema validado bajo los estándares de las buenas prácticas de automatización GAMP 5. Nuestra solución cumple con los requerimientos establecidos por la regulación CFR21 para registros y firmas electrónicas.
Entre los principales beneficios de eADMxL encontramos:
- Acceso controlado
- Perfiles de acceso
- Creación de registros (proyectos)
- Módulos de pruebas analíticas
- Obtención de reportes analíticos
- Firmas electrónicas
- Pistas de auditoría
Te invitamos a que nos contactes para que nuestros expertos te hablen a profundidad sobre los demás beneficios de nuestra herramienta y te muestren en tiempo real el funcionamiento.
- 0
- Por Team deappharma

26, Jun 2025
Situación actual en procesos de validación de métodos analíticos
Antes de abordar el tema es necesario saber ¿Qué es la validación de métodos analíticos? y el impacto que tiene este proceso dentro de las buenas prácticas de laboratorio. Recordemos que la validación de métodos analíticos es la evidencia documentada de que un método cumple con el propósito para el que fue diseñado ¿Pero que es evidencia documentada? Evidencia documentada hace referencia a la obtención de resultados de las pruebas analíticas que incluyen estadísticas tanto descriptivas como inferenciales mostrando con esto que el método en cuestión es capaz de ser; especifico, selectivo, lineal, preciso, exacto, reproducible y robusto cumpliendo con los requisitos de diseño preestablecidos. En la actualidad, este proceso solo se enfoca en la parte técnica. Es decir, solo en el entregable documental final dejando en segundo plano lo relacionado a la integridad de datos de los registros.
La manera común de reporteo y obtención de resultados para el proceso de validación de métodos analíticos dentro de la industria farmacéutica es mediante el uso de hojas de cálculo de Excel, mayormente. Excel, es una herramienta utilizada para captura, proceso y obtención de resultados. Sin embargo, esta no es la mejor práctica cuando de integridad de datos se trata. El uso de Excel para el procesamiento de datos para validación de métodos analíticos manifiesta problemas de cumplimiento debido a que estos archivos no se encuentran calificados o no cumplen con los controles de seguridad, trazabilidad, acceso y auditoría que la normatividad solicita. Recordemos que un dato integro es aquel que se puede mantener perdurable y por ende es: atribuible, legible, contemporáneo y exacto (ALCOA) a través del tiempo. Para poder alcanzar el objetivo de integridad de datos en procesos analíticos se requieren controles y fases específicas que brinden certidumbre regulatoria. Dichas fases y controles los podemos resumir a continuación:
- (Fase 1) Calificación de hojas de cálculo. Este rubro lo podemos denominar como “fase 1”, en la cual se realiza el proceso de evaluación y obtención de evidencia documentada para las etapas de calificación de diseño, instalación, operación y desempeño de las hojas de cálculo.
- (Fase 2) Mantenimiento y trazabilidad. Este rubro lo podemos definir como “fase 2”, en la cual se debe gestionar la hoja de cálculo calificada con atributos de integridad de datos. Es decir, dotar a los archivos de Excel con propiedades de acceso, permisos, protección, gestión, trazabilidad, firma electrónica y pistas de auditoría.
Recordemos que los controles de seguridad para las hojas de cálculo en las fases de mantenimiento y trazabilidad deben ser robustos. No es recomendable que la configuración de controles de seguridad y trazabilidad de hojas de cálculo se realice con herramientas de programación poco robustas como lo es VBA (Visual Basic Application). VBA manifiesta diferentes vulnerabilidades que ponen en riesgo un mantenimiento efectivo de hojas de cálculo utilizadas en los procesos analíticos. Las vulnerabilidades van desde el desbloqueo por herramienta de internet, colocación de pistas de auditoria dentro de los mismos libros de Excel y acceso controlados mediante contraseñas que no cumplen con los parámetros de seguridad solicitado por la guía CFR21 lo cual manifiesta un alto riesgo para la integridad de datos.
Es importante, tener presente que el proceso de validación de métodos analíticos no es una actividad aislada. Estas actividades tienen repercusión en diferentes áreas que dependen de la integridad de datos obtenidos durante el proceso. Las áreas que se pueden ver afectadas por hallazgos de auditoria por temas relacionados con la integridad de datos en métodos son aquellas como; calidad, validación de procesos, aseguramiento calidad y cumplimiento regulatorio por mencionar algunas.
En este articulo logramos analizar la situación actual del proceso de validación de métodos analíticos. ¿Pero existe solución al uso no controlado de Excel? ¿Qué alternativas podemos tener para dar cumplimiento a proceso de CSV de estos archivos? De primera instancia el proceso seguirá sin cumplimiento siempre y cuando no se tome acción. Y la acción la podemos definir bajo lo siguiente:
- Proyectos de calificación. Corresponde a realizar la calificación de cada hoja y libro de Exel utilizado en los procesos analíticos. Sin embargo, ante un gran número de archivos esta práctica tiende a ser poco sostenible tanto en costo como en tiempo de ejecución.
- Implementación de herramientas avanzadas como eADMxL. La implementación de herramientas de software que estén lógicamente construidas ayuda a robustecer el proceso analítico sin necesidad de crear, editar y reutilizar hojas de cálculo elevando con esto la integridad de datos. Sin duda, el uso de herramientas avanzadas son una excelente opción para potenciar y mitigar riesgos por el uso no controlado de hojas de cálculo en procesos de validación de métodos.
Recuerda que la integridad de datos no es opcional, este atributo contribuye con la calidad de tus productos. Te invitamos a transformar tu proceso analítico con nuestra herramienta avanzada, eADMxL, para el control de validación de métodos analíticos. eADMxL, te brinda un proceso lógico ayudándote a desprenderte de un número considerable de hojas de cálculo. Conoce los beneficios y alcance aquí>>eADMxL
Comprueba porque nuestra herramienta ya es utilizada en los mejores laboratorios de México y Latina.
Contáctanos y confirma como nuestras soluciones se adaptan a tus necesidades. ¡Da clic a qui para contactar!
¡Gracias por leernos!
3, Dic 2024
Modernice los procesos de validación de métodos analíticos para optimizar la integridad de datos
La industria farmacéutica está estructurada por múltiples procesos GxP. Dentro de estos procesos existe uno que se involucra directamente con los métodos analíticos para la cuantificación de sustancias activas, conservadores, productos de degradación y sustancias relacionadas contenidas en diferentes formas farmacéuticas. Este proceso se llama validación de métodos analíticos. El abordar métodos analíticos no es una tarea fácil, se necita pericia, enfoque analítico y procesos bien estructurados para la óptima obtención de resultados íntegros.
¿Como se aborda este proceso analítico? El proceso analítico de manera general se aborda por dos vías;
- (a) Verificación de método analítico. Este proceso parte de una implementación y posterior validación de método analítico bajo las condiciones descritas en farmacopeas nacionales y/o internacionales reconocidas internacionalmente. ¡Importante! La implementación de un método reconocido no deja exento al método de ser sometido a pruebas de validación analítica. El único beneficio que podemos encontrar en esta fase es la propuesta de un método base. Esperando que este funcione tal y como se describe en la referencia. De lo contrario este deberá ser actualizado y detallado bajo las condiciones específicas del laboratorio.
- (b)Validación de método analítico “In-House”. Este proceso parte del desarrollo de un método analítico bajo un conocimiento “Know-How” directamente relacionado a la capacidad analítica del laboratorio. Regularmente, esto sucede porque no hay métodos referentes en farmacopeas autorizadas.
No obstante, en cualquiera de los casos descritos con anterioridad, se debe considerar el procesamiento de datos y obtención de resultados en concordancia con los requisitos necesarios para dar cumplimiento a la integridad de datos. Recordemos que la integridad de datos es una manifestación consistente de un dato. Es decir, el dato debe perdurar, de tal forma que este pueda mantenerse; atribuible, legible, contemporáneo y exacto a través del tiempo. Regulatoriamente, el término integridad de datos no es nuevo. Sin embargo, en las actualizaciones más recientes de las normativas, este concepto ha tomado gran fuerza y relevancia dentro de los procesos GxP (BxP).
En la actualidad un gran número de laboratorios farmacéuticos utiliza para el proceso de validación de métodos analíticos el uso de hojas de cálculo. Sin embargo, un gran número de estas hojas no se encuentran validadas y/o la validación existente es poco robusta. Es aquí, donde la integridad de datos comienza a fallar. Ya que, un gran número de laboratorios farmacéuticos utiliza estas hojas sin contar con el respectivo soporte documental de validación del sistema computarizado.
Estamos convencidos, que validar las hojas de cálculo utilizadas para los procesos analíticos de validación de métodos analíticos es una tarea desafiante y altamente costosa. Es por ello, que la implementación de herramientas especializadas como eADMxL es el mejor aliado para disminuir el riesgo del uso de hojas de cálculo no validadas.
¿Como eADMxL me ayuda a optimizar el proceso de validación de métodos analíticos?
eADMxL es un complemento sistemático y lógico. En el cual, solo deberás capturar tus datos crudos para la obtención de resultados. La herramienta, cumple con los requisitos de integridad de datos. Ya que, cada dato ingresado en el sistema es contralado mediante; accesos, perfiles usuarios, módulos de análisis (parámetros de validación necesarios para pruebas de; valoración, impurezas, uniformidad de contenido y disolución), firmas electrónicas y pistas de auditoría por cada edición de dato crudo.
Te invitamos a conocer eADMxL y por qué esta herramienta ya está en los mejores laboratorios de habla hispana. ¡Solicita y agenda tu demostración aquí!
Contáctanos y solicita una demostración y asesoría gratuita ¡Quiero un asesoría y demostración!
¡Gracias por leernos!
21, Abr 2023
Validación/Verificación de procedimientos analíticos enfoque EDQM
La directriz de la ICH sobre «Validación de procedimientos analíticos: Texto y Metodología» (Q2) constituye un análisis de las características de validación que deben tenerse en cuenta durante la validación de un procedimiento analítico (la directriz también se ha adoptado para los veterinarios durante el debate de la VICH). Se dirigen principalmente a la industria farmacéutica
indicando qué datos de validación deben proporcionarse en un expediente de solicitud. Estos datos deben demostrar que las pruebas y los criterios de aceptación propuestos están suficientemente controlados para garantizar una calidad reproducible de los productos en el momento de su comercialización y un control adecuado durante su vida útil (estabilidad).
Dado que las circunstancias en las que trabaja una OMCL son diferentes de las de una empresa farmacéutica – en la mayoría de los casos no se realizan análisis rutinarios, sino que a menudo las respuestas deben en un corto periodo de tiempo, es necesario reconsiderar el grado de validación/verificación antes de realizar un análisis. Por otra parte, en todos casos debe garantizarse que el resultado presentado es fiable. También hay que destacar que los materiales de referencia adecuados son un factor importante tanto para la realización de los los estudios de validación/verificación como en el propio análisis. El uso de preparados de referencia es ampliamente aceptado puede evitar en determinadas circunstancias la consideración de algunas
características de validación, sobre todo en el ámbito de los productos biológicos: esto debe justificarse caso por caso. El OMCL podrá decidir el alcance necesario teniendo en cuenta los factores de riesgo.
El ámbito de aplicación de este documento -dirigido específicamente a los OMCL- es orientar sobre el alcance de la validación/verificación necesaria, en función de diversas circunstancias, es decir, el objetivo del análisis (por ejemplo, detección de incumplimiento), la cantidad de datos de validación ya disponibles (por ejemplo, en caso de transferencia de un método), la experiencia o los datos históricos ya disponibles en el OMCL individual (por ejemplo, recuperación a partir de una matriz compleja; uso rutinario de una valoración estándar incluso si se valoran sustancias diferentes), etc.
Este documento es igualmente aplicable a productos de origen sintético y biológico. No aborda las prácticas habituales de laboratorio: por ejemplo, orientaciones relativas al uso del equipo, calibración, etc.
Este documento es una nota orientativa que ofrece recomendaciones detalladas sobre el de validación/verificación en función de la categoría del procedimiento analítico; cabe señalar que siempre son posibles otros enfoques. Con respecto a la nueva Directiva 2010/63/UE relativa a la utilización ética de los animales para fines científicos y educativos y el convenio europeo sobre protección de los animales vertebrados utilizados para experimentación y Otros Fines Científicos (Consejo de Europa) es necesario prestar especial atención al uso de métodos in vivo como procedimientos analíticos. Debe hacerse todo lo posible para racionalizar y racionalizar y restringir al mínimo necesario la utilización de animales sobre la base de un análisis exhaustivo de la situación.
Cabe destacar que este documento no puede ofrecer asesoramiento detallado para todos los casos posibles en los que se utilicen ensayos in vivo. El propósito de este documento es proporcionar una orientación general. En todos los casos, deberá incluirse una breve descripción y/o justificación del enfoque elegido, incluidos los métodos, deberá describirse en la documentación interna del análisis.
Datos de validación
Deberán justificarse las modificaciones del método validado original. En función de la naturaleza de las modificaciones y del resultado de la evaluación del riesgo, podrán emprenderse actividades de validación o verificación suplementarias. Se aplicarán las mismas definiciones que en el documento ICH.
Categorías de análisis
En este capítulo se definen las diferentes situaciones analíticas (categorías) que pueden darse en un OMCL y las características de validación correspondientes que deben tenerse en cuenta. Consulte la versión actual de la directriz ICH sobre «Validación de procedimientos analíticos»: Text and Methodology» (Q2).
Los estudios formales de validación, de acuerdo con los requisitos de la ICH, deben llevarse a cabo cuando se desarrolle un nuevo método, cuando se utilice un método existente o cuando los datos de validación de un método existente deban completarse.
Según la norma ISO 17025, la validación es necesaria para los métodos no normalizados. .En el contexto de la OMCL, los métodos de farmacopea y los métodos validados de una autorización de comercialización se consideran métodos estándar.
La verificación del método debe realizarse para demostrar que en las condiciones reales de uso en los laboratorios individuales el método (validado) es adecuado (apto para su uso). Esto puede lograrse realizando las pruebas de idoneidad del sistema (por ejemplo, la resolución en un método cromatográfico) el control de la sensibilidad en el umbral de notificación, el control de la integridad de un paso de reacción (por ejemplo, extracción, reacción de hidrólisis) antes de que pueda realizarse la determinación propiamente dicha, verificar la precisión del método, etc. Esto también puede lograrse realizando un ejercicio de transferencia del método, en el laboratorio que ha establecido el método y el OMCL y comparando los resultados para demostrar la equivalencia. En todos los casos, se incluirá una breve nota en la que se expliquen los motivos del método elegido (en función de la complejidad del análisis requerido). La complejidad del análisis requerido-, deberá incluirse en la documentación interna del análisis. Las desviaciones de esta directriz deberán justificarse.
En las tablas 1 a 8 se contemplan varias categorías de análisis:
Tabla 1: Método publicado en la Farmacopea Europea
Los procedimientos analíticos descritos en una monografía se consideran validados. El OMCL debe verificar que todos los materiales de referencia necesarios y que se realizan las pruebas de idoneidad del sistema requeridas. Para las pruebas de sustancias relacionadas, la especificidad para cualquier impureza conocida que no figure en la monografía (por ejemplo, la lista de transparencia Ph Eur). Para las monografías de productos terminados, el OMCL debe verificar que ningún excipiente interfiera en el análisis de la sustancia activa, a menos que se indique contrario en la monografía.
Nota: Para entrar en esta categoría, los procedimientos deben ser descritos en detalle, no por ejemplo como en algunos casos para biológicos donde hay sólo una descripción general del método. Los detalles pueden proceder del informe publicado del estudio colaborativo (por ejemplo, los informes de estudios BSP en Pharmeuropa Bio & Scientific notes)
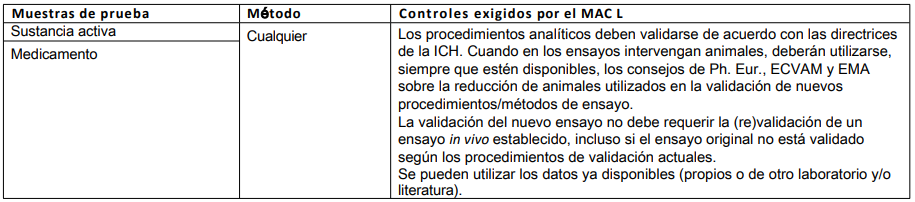
Tabla 2: Método validado de un fabricante (1ª autorización de comercialización)
Los procedimientos analíticos tomados de una autorización de comercialización están totalmente validados por la empresa.
1er MAH = producto fabricado por el MAH que validó el método original utilizado. 2º HAC = producto de un fabricante diferente
para el que no se ha validado específicamente el método original utilizado.
Cuando los métodos procedan de un expediente o expedientes de solicitud antiguos sin datos de validación o con datos insuficientes, se deberá informar a la Autoridad Competente supervisora. Para las características de validación que deben tenerse en cuenta, véanse las tablas 2, 5.
Tabla 3: Método publicado no compendial
Las características de validación a considerar dependerán siempre de la cantidad de datos de validación aportados. Si el método ha sido completamente validado y los datos se han publicado en la bibliografía, véase el cuadro 1. En caso contrario, deberá tenerse en cuenta lo siguiente.
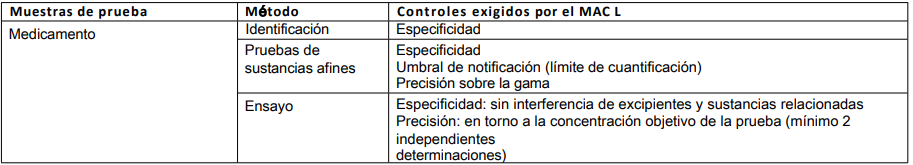
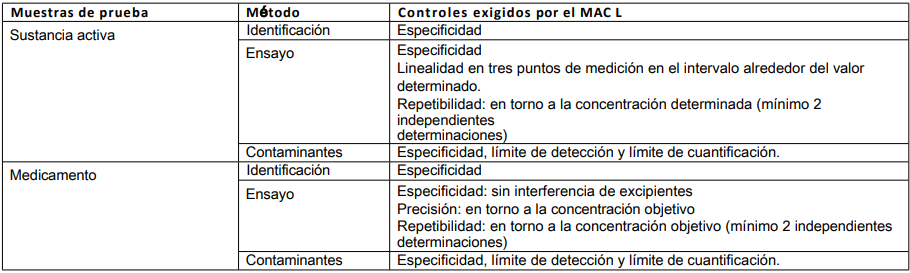
Tabla 4: Método de sustancia activa utilizado para un medicamento
El principal factor a considerar aquí es la influencia de la matriz en el análisis, incluyendo la interferencia de los excipientes.
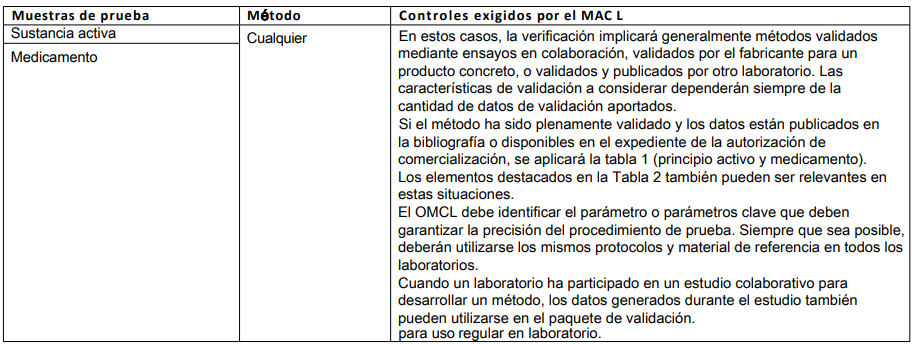
Tabla 5: Métodos validados para reducir, perfeccionar o sustituir el uso de animales (3R)
Tabla 6: Detección de incumplimientos
El cribado de la no conformidad significa que el objetivo del análisis es detectar la posible no conformidad del producto con las especificaciones.
Este tipo de cribado se realizaría cuando se solicita un análisis rápido y/o cuando no se dispone de datos de validación del método. El procedimiento debe documentarse en todos los casos.
Si se detecta un incumplimiento, debe ampliarse el alcance de la validación, por ejemplo, considerando la posibilidad de cambiar a un método bien reconocido (método de compendio o método del expediente MAH).
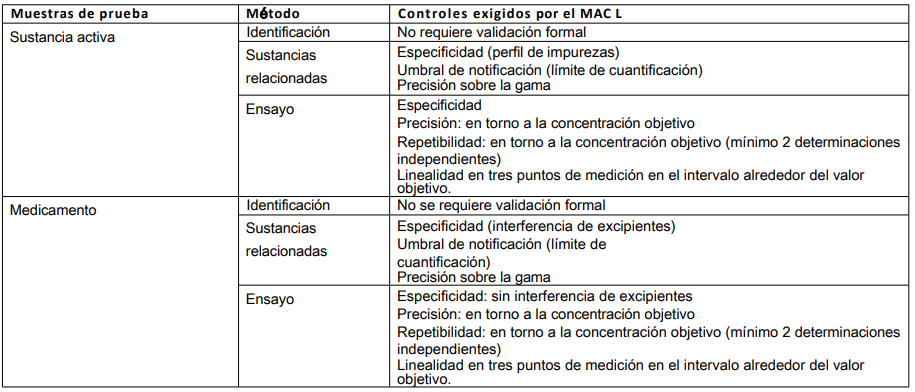
Tabla 7: Detección de productos/contaminantes desconocidos
En estos casos, se carece de información sobre el producto que debe analizarse con respecto a su declaración en la etiqueta (presencia o ausencia de determinadas sustancias) o para aclarar otros aspectos solicitados por la Inspección.
Pruebas a tener en cuenta: identificación, ensayo y, tal vez, pruebas de pureza. El primer paso importante es identificar los principales componentes del producto.
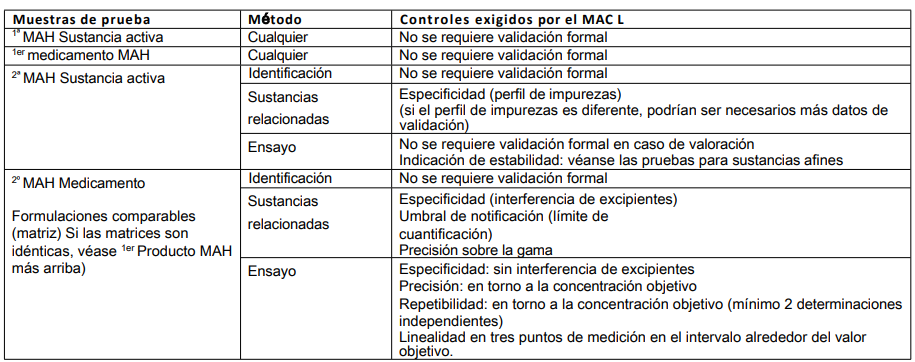
Tabla 8: Desarrollo de un nuevo método
Esto ocurre principalmente cuando un producto se prueba en condiciones de ensayo rutinarias y/o cuando se utiliza un procedimiento analítico interno.

¿Existen herramientas en el mercado que mejoren los procesos administrativos y de laboratorio?
Como bien sabemos el desafío más grande al que se a puesto a prueba el sector farmacéutico es el relacionado con la integridad de datos. Y el proceso de validación de método analíticos no cae fuera de este requerimiento ¡Pero! ¿Por qué es tan complejo este tema? La respuesta se deriva por la gran cantidad de datos y documentos que se emiten día con día en el que se ven impactados documentos y libros de Excel.
Recordemos que la validación de software y libros de Excel es el primer paso. No obstante, muchos libros de Excel utilizados en el sector farmacéutico no cuentan con evidencia de validación lo cual es un riesgo que podrá derivar en un hallazgo critico ante una inspección regulatoria.
El segundo paso y el más complejo es adherir las configuraciones necesarias que eleven la integridad de datos y favorezcan el mantenimiento tanto de software como de Libros de Excel.
Te invitamos a mitigar el riesgo por el uso de sistemas computarizados no validados y con pobres atributos de integridad de datos. Nuestros desarrollos están estructurados bajo un sistema de gestión de calidad trazable y auditable. Tales cuales cuentan con el soporte de validación-calificación documental con base en Gamp 5 y CFR21 parte 11.
Conoce eADMxL el cual es un software que desarrollamos para mitigar el uso de hojas de cálculo para procesos de validación de métodos analíticos. Conoce sus beneficios y alcance general aquí: ¡Da clic aquí!
“No vivas con el riesgo ¡Mitígalo!” para eso nosotros de ayudamos.
Visita nuestro sitio www.deappharma.com y solicita una demostración y asesoría gratuita.
¡Contáctanos! y conoce de que manera te vamos a ayudar.
¡Únete a nuestra comunidad en redes!
Referencias
- General European OMC L Network (GEON) Quality Management Document
- Validation of analitycal procedures :text and methodology Q2
6, Mar 2023
Importancia de la integridad de datos en procesos de validación de métodos analíticos
La integridad de datos es un aspecto crucial en el proceso de validación de métodos analíticos en la industria farmacéutica. La validación de métodos analíticos es un proceso que asegura que un método analítico específico es adecuado para su uso previsto y que proporciona resultados precisos y confiables.
La integridad de los datos es esencial en la validación de métodos analíticos porque los datos generados durante el proceso de validación se utilizan para establecer la precisión, la exactitud, la linealidad, la especificidad y la robustez del método analítico. La falta de integridad de los datos puede comprometer la precisión y confiabilidad de los resultados y poner en riesgo la seguridad y eficacia de los productos farmacéuticos.
Además, las autoridades reguladoras exigen que los datos utilizados en la validación de métodos analíticos sean precisos, confiables y completos. La falta de integridad de los datos puede poner en duda la validez de los resultados de la validación y poner en riesgo la aprobación de los productos.
Por lo tanto, la integridad de los datos es fundamental en el proceso de validación de métodos analíticos y se deben implementar controles rigurosos para garantizar que los datos generados durante el proceso de validación sean precisos, confiables y completos. Esto incluye la capacitación del personal, la implementación de controles de acceso y seguridad, la validación de sistemas, la verificación de datos y la revisión regular de la documentación y registros.
¿Cuales son los 5 hallazgos de integridad de datos en Hojas de cálculo en el sector farmacéutico?
A continuación de algunos hallazgos comunes de integridad de datos que se han observado en hojas de cálculo en sectores regulados, incluido el farmacéutico:
- Errores de entrada de datos: Los errores de entrada de datos son comunes en las hojas de cálculo y pueden deberse a errores humanos al ingresar información. Estos errores pueden incluir errores tipográficos, errores de transposición o errores de formato.
- Fórmulas incorrectas: Las fórmulas incorrectas pueden llevar a resultados inexactos y pueden deberse a una variedad de factores, como errores humanos, cambios en los datos de origen y copiar y pegar incorrectos.
- Control de cambios insuficiente: La falta de control de cambios puede hacer que sea difícil realizar un seguimiento de los cambios realizados en una hoja de cálculo y puede dificultar la identificación de errores o fraudes.
- Falta de validación de datos: La falta de validación de datos puede hacer que los datos sean inexactos o incompletos. Esto puede incluir la falta de validación de datos de entrada o la falta de validación de datos calculados por fórmulas.
- Falta de documentación: La falta de documentación puede dificultar la comprensión de cómo se creó una hoja de cálculo, lo que puede hacer que sea difícil identificar errores o fraudes. Es importante mantener una documentación completa y actualizada de las hojas de cálculo para garantizar la integridad de los datos.
Ahora que conoces la importancia de la integridad de datos en el proceso de validación de métodos analíticos y los hallazgos recurrentes en la industria vamos con el siguiente paso ¡Mejorar el proceso! ¿Quieres saber como?
¡DA CLIC AQUI!

¡Contáctanos! y conoce de que manera te vamos a ayudar.
¡Únete a nuestra comunidad en redes!
31, Ene 2023
Validación de métodos analíticos
Validación en la industria
Validación se puede definir como “encontrar o evaluar la verdad de algo”. Cuando un método analítico es usado para generar resultados acerca de las características de la sustancia de interés (eg., Api o medicamentos), es vital que los resultados sean confiables, ya que estos serán usados como base para la toma de decisiones en relación, con la definición de la forma de administración del fármaco o medicamento al paciente.
Un estudio de validación, el cual deriva en un método analítico tiene como finalidad el asegurar que los resultados obtenidos sean fidedignos (ALCOA ++) siempre que estos, sean obtenidos dentro del proceso analítico.
La validación de métodos analíticos es solo uno de los tipos de validación requerida durante el desarrollo y/o manufactura del producto. Para cumplir con los requerimientos de las buenas prácticas de manufactura (GMP), las compañías farmacéuticas deberán tener una política general de validación, en la cual se detalle que documentación, parámetros y evaluación, serán los necesarios para desempeñar dicha acción, sin dejar de considerar como alcance las validaciones correspondientes a : Procesos de producción, procedimientos de limpieza, métodos analíticos, evaluación y pruebas de control dentro del proceso , así como , los sistemas computarizados.
El objetivo y razón de incluir la validación como un requerimiento en la cGMP y en la NOM 059 (Regulación local), es de asegurar la calidad en cada paso de la fabricación del fármaco o medicamento, y que la calidad no solo sea evaluada bajo un análisis final del producto. La validación está destinada a proveer el aseguramiento de la calidad de un proceso o sistema, y la cual ha sido establecida, diseñada para derivar a la redacción de una metodología de calidad, para ser usada en un análisis de control de; Proceso, liberación y/o estudios de estabilidad.
El aseguramiento de la calidad de un medicamento, no se encuentra solamente por un simple análisis, si no tal y como se mencionó, es el conjunto de análisis con su respectiva y previa documentación, que den soporte de validación en cada etapa del proceso (ALCOA ++ durante toda la cadena de suministro).
Necesidad de la validación
Cada día, se realizan en el mundo un alto número de análisis, relacionados al monitoreo de compuestos orgánicos, y estas mediciones son utilizadas en algunas situaciones para la toma de decisiones, en áreas como son; control de calidad durante el proceso y la manufactura, comercios, y consumo. Entonces, muchas de estas decisiones, están con base en las mediciones o resultados obtenidos, como son; Liberación de un lote, liberación de producto, lanzamiento de un producto, que al final de la cadena de suministro puede afectar la salud, reputación, costos de análisis, retrabajos y un impacto directo económico, si no se evalúa correctamente un dato. Es aquí donde, la evaluación y obtención de un dato reproducible y seguro (Data Integrity) muestra su importancia, por estas razones, los requerimientos de los laboratorios incluyeron el termino de validación de métodos dentro de sus procesos analíticos.
Los governance de concientización han sido establecido como un requerimiento de control de calidad creado por agencias regulatorias para verificar que los laboratorios están correctamente desempeñando este trabajo, grupos de trabajo regulatorio desarrollan guías oficiales para llevar a cabo la validación, así como el establecimiento de sus criterios. Estas referencias (guías) deberían deberán ser evaluadas para garantizar con esto la confianza.
Los métodos analíticos deben ser validados por profesionistas analistas. Un método analítico con poca confianza para evaluar ensayos muy seguramente provea datos falsos, y entonces esto podría desencadenar en un retiro de producto (Recall).
El ejercicio de validación es costoso y consume mucho tiempo. Normalmente esta función es desarrollada por un área de calidad o soporte analítico, lo que puede alterar el trabajo normal del laboratorio. Sin embargo, el uso de métodos validados elimina repeticiones en el laboratorio y mejora el prestigio de laboratorio favoreciendo a los clientes y la rentabilidad a largo plazo.
En resumen, la validación de un método analítico deberá ser desempeñado por las siguientes razones:
- Elevar la integridad y calidad de los resultados.
- Los productos podrán alcanzar la aceptación por agencias internacionales.
- Lograr (cuando aplique) un rango de aprobación como método “oficial/método de referencia” por las agencias regulatorias.
- Te apoya a lograr la acreditación de requerimientos mandatorios para laboratorios por las guías ISO 17025 y requerimientos normativos GxP.
- Mejora el balance financiero del laboratorio.
Propósito de la validación
Un método analítico detalla los pasos necesarios a desempeñar en un análisis, los cuales pueden incluir, pero no limitados a la descripción de; Preparación de las muestras, estándares y reactivos, uso de algún aparato o instrumento, generación de una curva de calibración, uso fórmulas para realizar el cálculo, etc. El propósito de la validación de un método analítico debe ser adecuado, porque de esto depende el diseño hacia el uso previsto para el que será utilizado.
El uso de los métodos analíticos durante etapas del desarrollo del producto y/o manufactura, provee información con relación a lo siguiente y de los cual depende el alcance de validación:
- Potencia, La cual puede estar correlacionada de manera directa a los requerimientos de una dosis conocida o al establecimiento de esta.
- Impurezas y/o sustancias relacionadas, cuales pueden estar relacionados a mostrar el perfil de seguridad impurezas, la interpretación y predicción en la formación de estas (se asocia directamente con el paciente).
- Evaluación de las características claves del fármaco (caracterización), como son; forma cristalina (polimorfismo), liberación del fármaco (forma farmacéutica de dosificación), uniformidad y disolución (pruebas de aptitud de proceso esenciales para pruebas de liberación), las cuales, al ser evaluadas nos dan información sobre las propiedades que pueden comprometer la biodisponibilidad.
- Productos de degradación, los métodos necesitan ser indicativos de estabilidad, siempre y cuando estos sean utilizados en estudios de estabilidad de medicamentos y fármacos (esencial uso de PDA o MS-LC para una correcta evaluación de la pureza cromatográfica y pureza química). Es indispensable que estos productos de degradación estén cuantificados dentro de un umbral adecuado (establecer el límite con base a ICH Q3A (R) e ICH Q3B (R)) que no sea un riesgo para el paciente, las guías anteriormente mencionadas marcan la directriz específicamente para las impurezas que pueden surgir como productos de degradación de la sustancia farmacológica, o que surgen de interacciones entre la sustancia farmacológica y los excipientes o componentes de los materiales de envasado primarios. La directriz establece una justificación para la notificación, identificación y calificación(cuantificación) de tales impurezas, en base a una evaluación científica de las impurezas probables y reales observadas, y de las implicaciones de seguridad, siguiendo los principios elaborados en la directriz principal. Estas guías describen y proponen valores umbral para informar y controlar las impurezas, en función de la dosis diaria máxima de la sustancia farmacológica administrada en el producto.
Los parámetros y efectos descritos anteriormente, así como su buena implementación aseguran que la producción de la sustancia fármaco o medicamento sean consistente.
La validación de métodos analíticos entonces; es un proceso relevante y este debe ser desempeñado. Ya que este proceso genera los datos necesarios para demostrar integridad, confiabilidad y consistencia. Con base en todo el análisis previó, entonces, podemos definir que el propósito de realizar o desempeñar la validación analítica es demostrar que los procesos involucrados en el desarrollo y manufactura del producto, tales cuales son; producción, limpieza y evaluación analítica, pueden ser desempeñadas de una manera efectiva y reproducible y con esto generan calidad, seguridad y eficacia.
Ahora que conoces la la importancia del proceso de validación vamos con el siguiente paso ¡Mejorar el proceso! ¿Quieres saber como?
¡DA CLIC AQUI!
¡Contáctanos! y conoce de que manera te vamos a ayudar.
¡Únete a nuestra comunidad en redes!
20, May 2022
Linealidad del sistema
Es la capacidad del método analítico para obtener resultados directamente proporcionales a la concentración o cantidad del analito en un rango definido.
La linealidad puede demostrarse directamente en la sustancia farmacológica (mediante dilución de una solución madre estándar) y/o pesadas separadas de mezclas sintéticas de los componentes del producto farmacéutico, utilizando el procedimiento propuesto. Este último aspecto puede estudiarse durante la investigación en el rango definido. La linealidad debe evaluarse mediante la inspección visual de un gráfico de señales en función de la concentración o el contenido del analito. Si existe una relación lineal, los resultados del ensayo deben evaluarse mediante métodos estadísticos apropiados, por ejemplo, mediante el cálculo de una línea de regresión por el método de los mínimos cuadrados. En algunos casos, para obtener la linealidad entre los ensayos y las concentraciones de la muestra, puede ser necesario someter los datos del ensayo a una transformación matemática antes del análisis de regresión. Los datos de la línea de regresión pueden ser útiles para proporcionar estimaciones matemáticas del grado de linealidad.
Deben presentarse valores tales como:
Coeficiente de correlación (r). Muchos autores planteanque para que el método se considere lineal, el coeficiente de correlación debe ser mayor que 0,999. Sin embargo, la mejor forma de indicar la linealidad del método estudiado será realizar una prueba estadística de t (t de Student), en la cual se calculará la correlación lineal significativa (tr) a partir de la hipótesis nula de no correlación entre las magnitudes estudiadas («x» y «y«).
Pendiente (conocida también como coeficiente de regresión). Indica la sensibilidad de calibración o del método y se expresa en unidades de respuesta sobre unidades de concentración o cantidad del analito. La sensibilidad analítica relaciona la aleatoriedad de la respuesta con la aleatoriedad debida a la variación de la concentración, es inversamente proporcional a la capacidad de detectar pequeñas diferencias en la concentración del analito, y se obtiene dividiendo la pendiente de la curva de calibración por la desviación estándar de las respuestas en cada punto o concentración. Se considera que, a mayor pendiente, mayor sensibilidad y que mientras más pequeño sea el coeficiente de variación de la pendiente mayor será la linealidad (coeficientes de variación de la pendiente mayores que el 5,0 % indican falta de linealidad).
Intercepto. Es el estimador que se relaciona con la presencia de interferencias o errores sistemáticos. El intervalo de confianza del intercepto debe incluir al cero para cumplir con el requisito de proporcionalidad (como se exige para el cumplimiento de la ley de Lambert-Beer en los métodos espectrofotométricos). Se determinará la prueba de proporcionalidad mediante una prueba t considerando como hipótesis nula que el intercepto tiene que ser cero.
La intersección y la pendiente de la línea de regresión, así como la suma residual de los cuadrados. Debe incluirse un gráfico de los datos (reporte). Además, deberá presentarse un análisis de la desviación de los puntos de datos reales con respecto a la línea de regresión puede ser útil para evaluar la linealidad.
Algunos procedimientos analíticos, como los inmunoensayos, no demuestran linealidad
después de cualquier transformación. En este caso, la respuesta analítica debe describirse mediante una función apropiada de la concentración (cantidad) de un analito en una muestra. Para establecer la linealidad, se recomienda un mínimo de 5 concentraciones. Deberán justificarse otros enfoques.
Proceso de linealidad del sistema controlado y gestionado con eADMxL.
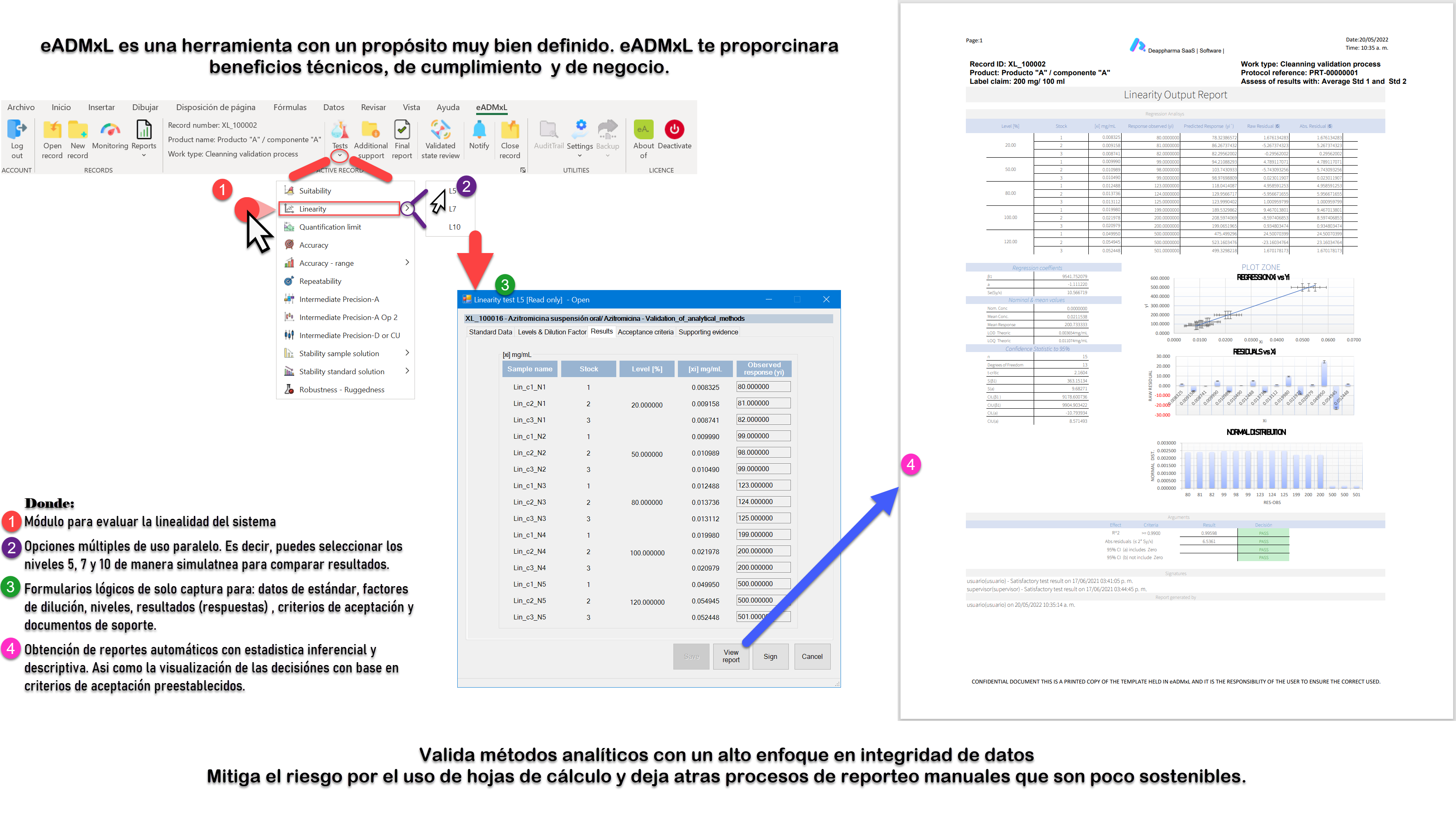 No olvides que, en caso de ser necesario, puedes contactarnos para mejorar el proceso de laboratorio y administrativo de tu organización a través software 100% confiable y seguro. Recuerda que en deappharma contamos con un software que te ayudara a controlar tu proceso de validación de métodos analíticos. Somos expertos en la materia, por lo que procesos complejos no representa un reto para nosotros, debido a que tenemos en mente los aspectos relevantes de cumplimiento.
No olvides que, en caso de ser necesario, puedes contactarnos para mejorar el proceso de laboratorio y administrativo de tu organización a través software 100% confiable y seguro. Recuerda que en deappharma contamos con un software que te ayudara a controlar tu proceso de validación de métodos analíticos. Somos expertos en la materia, por lo que procesos complejos no representa un reto para nosotros, debido a que tenemos en mente los aspectos relevantes de cumplimiento.
Referencias
- Rampazoo P. Standardisation and validation of analytical methods in the pharmaceutical industry. Il Farmaco 1990; 45:807-15.
- Comellas L. Desarrollo de métodos en HPLC. Barcelona: Instituto Químico de Sarria, 1994:68.
- VALIDATION OF ANALYTICAL PROCEDURES: TEXT AND METHODOLOGY Q2(R1)
¡Contáctanos! y conoce de que manera te vamos a ayudar.
¡Únete a nuestra comunidad en redes!
15, May 2022
Idoneidad del sistema (HPLC, MS-HPLC)
Las pruebas de idoneidad del sistema son una parte integral de muchos procedimientos analíticos. Las pruebas se basan en el concepto de que el equipo, la electrónica, las operaciones analíticas y las muestras a analizar constituyen un sistema integral que puede ser evaluado como tal. Los parámetros de las pruebas de idoneidad del sistema que deben establecerse para un procedimiento concreto dependen del tipo de procedimiento que se valide.
La evaluación de la aptitud del sistema se recomienda para todos los métodos analíticos, ya que permite verificar que el sistema de medición funciona apropiadamente, independientemente de las condiciones ambientales. Es conveniente que antes de llevar a cabo la validación del método analítico, se establezcan los criterios apropiados para la operación del sistema de medición, para ser evaluados en la validación y verificados de manera rutinaria al emplear el método analítico.
Métodos cromatográficos
La solución de aptitud del sistema se establece en el desarrollo del método y puede ser una solución del analito al 100 % del rango de linealidad o más concentrado. Puede ser una mezcla del analito con otro componente para verificar la resolución del analito y su(s) compuesto(s) de degradación o relacionados (ya sea agregándolos o degradando la muestra).
Metodología
La evaluación se recomienda con la inyección por sextuplicado la solución de aptitud del sistema. Calcular el Coeficiente de Variación (CV) del analito y se procede reportar.
- Factor de capacidad (K’)
- Resolución (R)
- Tailing ( T )
- Platos teóricos (N)
- Factor de coleo
Los valores mínimos y/o máximos para cada uno de los parámetros anteriores, son los siguientes:
- Factor de capacidad (K’) > 10 (recomendable)
- Coeficiente de variación (CV) ≤ 2% para proceso de valoración, disolución y ≤ 20% para procesos de evaluación de impurezas individuales.
- Resolución (R) > 1.5 (recomendable si se observa la presencia de productos de degradación y/o impurezas)
- Tailing ( T ) <1>
- Factor de coleo >2.5
Actualmente los procesos y evaluación de métodos analíticos por cromatografía liquida (HPLC, MS-HPLC etc..) establecen como parámetro de evaluación la idoneidad del sistema. Sin embargo como proceso integral de la validación de métodos analíticos esta prueba debe formar parte evaluada de este. De esta manera podemos obtener de manera verificable el inicio de las condiciones de validación.
Las pruebas de idoneidad del sistema son pruebas que deben ser evaluadas previamente a lanzar alguna corrida analítica, con el objetivo de verificar que las condiciones del sistema son optimas para llevar a cabo el análisis. La idoneidad del sistema siempre debe ser evaluada con soluciones de referencias, es decir estándares. No es sugerible que la idoneidad incluya la inyección y previsualización de resultados (áreas) de muestras. Ya que esta acción de inyección de muestras previamente a lanzar la corrida analítica, son consideradas actualmente malas practicas de laboratorio en lo asociado a integridad de datos.
Recordemos que la idoneidad del sistema debe formar parte integral del proceso de validación de manera inicial ya que esta es una buena referncias para determinar el momento de fallo y establecer limites de especificaciones vinculadas a este parametro de desempeño.
Proceso de idoneidad controlado y gestionado con eADMxL.
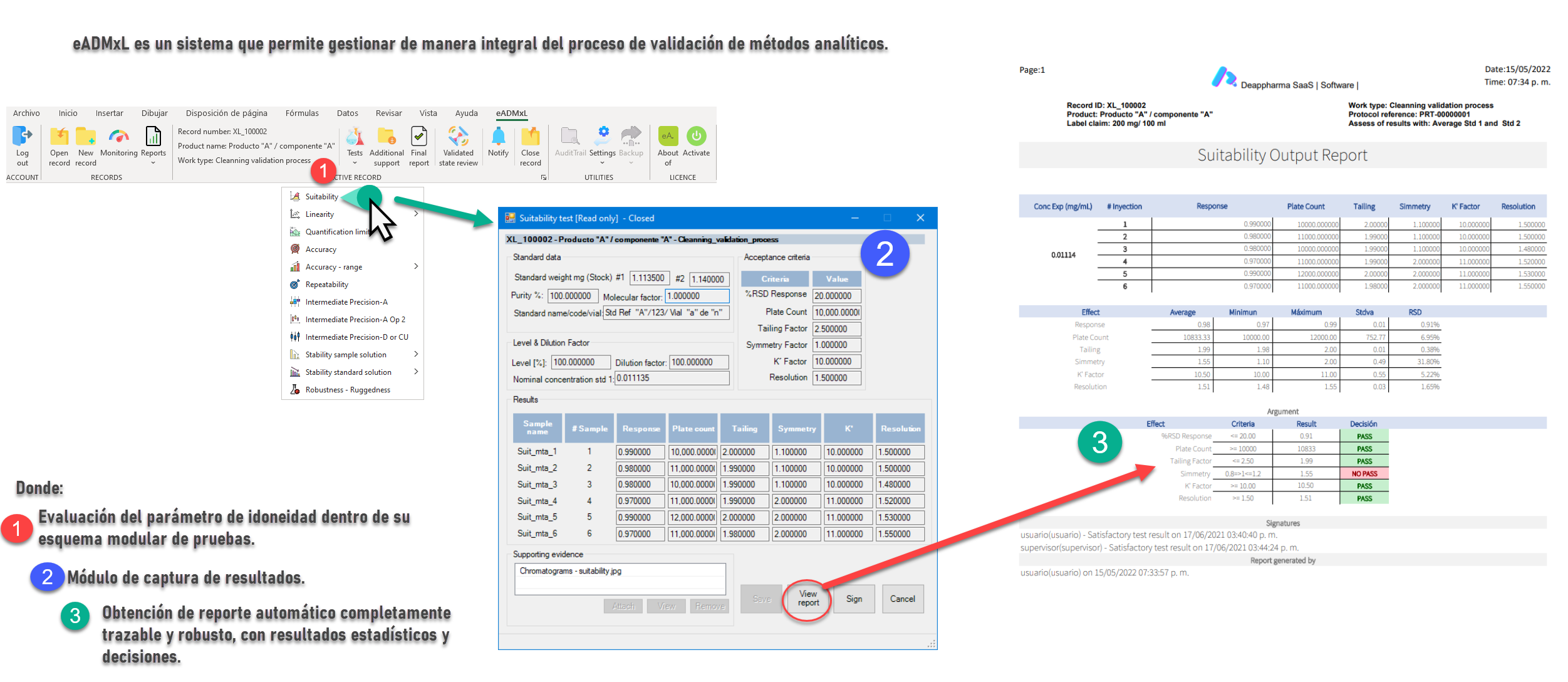
No olvides que, en caso de ser necesario, puedes contactarnos para mejorar el proceso de laboratorio y administrativo de tu organización a través software 100% confiable y seguro. Recuerda que en deappharma contamos con un software que te ayudara a controlar tu proceso de validación de métodos analíticos. Somos expertos en la materia, por lo que procesos complejos no representa un reto para nosotros, debido a que tenemos en mente los aspectos relevantes de cumplimiento.
Referencias
- VALIDATION OF ANALYTICAL PROCEDURES: TEXT AND METHODOLOGY Q2(R1)
¡Contáctanos! y conoce de que manera te vamos a ayudar.
¡Únete a nuestra comunidad en redes!
5, Abr 2022
ICH Q14 desarrollo de métodos analíticos y el impacto sobre ICH Q2 validación de métodos analíticos
Uno de los procesos GxP relevantes dentro de la industria es el proceso de validación de método analíticos. La importancia esencial es su vinculación directa con el método analítico utilizado de rutina (Liberaciones, Estabilidades). Con la actualización “propuesta” por parte de ICH, en lo referente a los procesos analíticos de desarrollo y validación se busca que estos tengan una relación estrecha en el que el beneficio principal es la calidad por diseño y mantenimiento del estado validado.
Como bien sabemos, el objetivo de la validación de procedimientos analíticos es demostrar que el procedimiento es adecuado para el fin previsto. Sin perder de vista que los datos y resultados deben ser suficientemente confiables para la toma de decisiones.
Un estudio de validación está diseñado para proporcionar pruebas suficientes de que el procedimiento analítico cumple sus objetivos. Estos objetivos se describen con un conjunto adecuado de características de rendimiento correspondientes, que pueden variar en función de uso previsto del procedimiento analítico y la tecnología especifica seleccionada.
Con la reciente propuesta de la guía ICHQ14 se busca incluir, describir y robustecer el proceso de laboratorio por la estrecha relación que existe entre el desarrollo de métodos y la validación de estos. Con esto podremos visualizar términos como son: estrategia de validación y control, ciclo de vida y mantenimiento como parte esencial de esta. Sin perder de vista la inclusión de metodologías existentes, tales cuales no eran claramente abordadas.
La propuesta ICH Q14 atribuye que dicho proceso y los conocimientos arrojados por este, se controlen de tal manera que se pueda obtener la información sobre la finalidad prevista del procedimiento analítico, y el rendimiento de las características y criterios asociados que deben ser validados.
La siguiente figura muestra cómo se puede generar el conocimiento durante el desarrollo de un procedimiento analítico propuesto en la ICHQ14 y con esto ayudar al diseño de un estudio de validación.
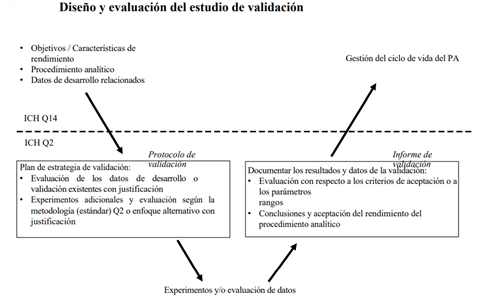
Validación durante el ciclo de vida de un procedimiento analítico
Durante el ciclo de vida de un procedimiento analítico pueden ser necesarios cambios. En tales casos, los cambios parciales pueden necesitar una revalidación completa. La ciencia y los principios basados en el riesgo pueden utilizarse para justificar sin una determinada característica de rendimiento necesita o no revalidación. El alcance de la revalidación depende de las características de rendimiento analítico afectadas por el cambio.
Consideraciones generales para el desarrollo de procedimientos analíticos y la gestión del ciclo de vida
El objetivo del desarrollo es obtener un procedimiento analítico adecuado para su propósito: medir un atributo o atributos del material analizado con la especificidad/selectividad, exactitud y/o precisión en el intervalo notificable. En esta sección se describen los enfoques mínimo y mejorado del desarrollo de procedimientos analíticos En esta sección se describen los enfoques mínimo y mejorado para el desarrollo de procedimientos analíticos. Aunque el enfoque mínimo sigue siendo aceptable, algunos o todos los elementos del enfoque mejorado de este enfoque mejorado para apoyar el desarrollo y la gestión del ciclo de vida de los procedimientos analíticos.
En ciertos casos, un procedimiento analítico establecido puede aplicarse a múltiples productos con poca o ninguna modificación de las condiciones de medición. Para una nueva aplicación de dichos procedimientos analíticos de plataforma plataforma, el desarrollo posterior puede abreviarse y algunas pruebas de validación puede omitirse sobre la base de una justificación científica y de riesgo. Los detalles de las características de rendimiento consideradas para la validación de procedimientos analíticos se describen en la ICH Q2.
En general, los datos obtenidos durante los estudios de desarrollo (por ejemplo, los datos de robustez de un diseño de de experimentos (estudio DoE)) pueden utilizarse como datos de validación para las características de rendimiento del procedimiento analítico características de rendimiento del procedimiento analítico y no es necesario repetirlos.
Enfoques mínimos frente a enfoques mejorados para el desarrollo de procedimientos analíticos
Enfoque mínimo.
El desarrollo de procedimientos analíticos debe incluir los siguientes elementos, según corresponda:
- Identificación de los atributos de la sustancia o el producto farmacéutico que deben ser analizados por el procedimiento analítico.
- Selección de una tecnología de procedimiento analítico apropiada y de los instrumentos o aparatos adecuados.
- Llevar a cabo estudios de desarrollo apropiados para evaluar el rendimiento del procedimiento analítico
- características de rendimiento del procedimiento analítico, como la especificidad, la exactitud y la precisión en el intervalo notificable (incluyendo el modelo de calibración, los límites en los extremos del rango inferior y/o superior) y la solidez.
- Definir una descripción adecuada del procedimiento analítico que incluya la estrategia de control estrategia de control del procedimiento analítico (por ejemplo, ajustes de los parámetros y adecuación del sistema).
Enfoque mejorado
El enfoque mejorado ofrece una forma sistemática de desarrollar y perfeccionar el conocimiento de un procedimiento analítico. Un enfoque mejorado debe incluir uno o más de los siguientes elementos, además de los ya descritos para el enfoque mínimo:
- Una evaluación de las propiedades de la muestra y de la variabilidad esperada de la muestra basada en conocimiento del proceso de fabricación.
- Definición del perfil analítico objetivo.
- Realización de una evaluación de riesgos y evaluación de los conocimientos previos para identificar los parámetros del procedimiento analítico que pueden afectar al rendimiento del procedimiento.
- Realización de experimentos uni o multivariados para explorar los rangos y las interacciones entre parámetros identificados del procedimiento analítico.
- Definir una estrategia de control del procedimiento analítico basada en una mejor comprensión del procedimiento incluyendo los puntos de ajuste y/o rangos apropiados para los parámetros relevantes del procedimiento analítico garantizando el cumplimiento de los criterios de rendimiento.
Definir un plan de gestión de cambios del ciclo de vida con definiciones claras y categorías de información de condiciones establecidas, rangos aceptables probados o regiones de diseño operacional del método según proceda. La aplicación de elementos del enfoque mejorado al desarrollo puede conducir a procedimientos analíticos más sólidos, a una mejor comprensión del impacto de los parámetros del procedimiento analítico y a una mayor flexibilidad para la gestión del ciclo de vida, como rangos operativos más amplios, un conjunto más apropiado de condiciones establecidas y categorías de información asociadas para los cambios.
El enfoque mejorado ofrece potencialmente varias ventajas, entre ellas
- Comprensión de aquellos atributos del procedimiento analítico tales cuales son esenciales para el rendimiento del procedimiento.
- Mejorar el control de los procedimientos analíticos para conseguir un funcionamiento más fiable.
- Permitir medidas preventivas y facilitar la mejora continua mediante el uso de más conocimiento de los procedimientos analíticos.
- Reducir la cantidad de esfuerzo a lo largo del ciclo de vida del procedimiento analítico.
El ciclo de vida del procedimiento analítico
La siguiente figura muestra los elementos del ciclo de vida del procedimiento analítico.
El desarrollo de procedimientos analíticos y los enfoques de gestión de cambios se describen en esta directriz, mientras que la validación de los procedimientos analíticos se describe en la ICH Q2. Dependiendo del uso previsto del procedimiento analítico y del enfoque de desarrollo adoptado, el orden y el alcance de cada elemento pueden variar, y varios elementos pueden darse simultáneamente.

Estrategia de control del procedimiento analítico
Una estrategia de control de procedimientos analíticos debe garantizar que el procedimiento analítico funcione como esperado durante su uso rutinario a lo largo de su ciclo de vida y consiste en un conjunto de controles, derivados de la comprensión actual del procedimiento analítico, incluidos los datos de desarrollo, la evaluación de riesgos y la robustez. El conocimiento previo también podría utilizarse para desarrollar la estrategia de control del procedimiento analítico.
La estrategia de control del procedimiento analítico debe definirse antes de la validación (ICH Q2) y debe confirmarse una vez finalizada la validación.
La estrategia de control del procedimiento analítico incluye los parámetros del procedimiento analítico que necesitan control y la prueba de idoneidad del sistema que forma parte de la descripción del procedimiento analítico. La descripción del procedimiento analítico debe incluir los pasos necesarios para realizar cada prueba analítica. Esto puede incluir (pero no se limita a) la muestra, los materiales de referencia y los reactivos, la preparación de la muestra y de control, el uso del aparato, la generación de la curva de calibración, el uso de las fórmulas con herramientas para el cálculo de los resultados notificables y otros pasos necesarios. El nivel de detalle debe permitir a un analista experto realizar el análisis e interpretar los resultados (como el nivel de detalle en una farmacopea regional para una sustancia similar). Esto se denomina comúnmente comprobación de la calidad de los datos. Se recomienda un seguimiento continuo de los resultados de los procedimientos analíticos seleccionados para buscar cualquier tendencia, de acuerdo con las expectativas. La revisión de los resultados de los procedimientos analíticos facilita la gestión del ciclo de vida del procedimiento y permite una intervención proactiva para evitar fallos.
El desafío en la revisión y mantenimiento del estado validado
La directriz ICH 14 establece de manera concisa la relación que existe entre el desarrollo de métodos, el proceso de validación y el mantenimiento del ciclo de vida con un enfoque en Calidad. Sin embargo, uno de los desafíos que enfrentamos en el día a día y llegado el momento es aquella que relaciona la obtención, disponibilidad y compilación de la información para realizar la revisión de estado validado. Sin duda la estructuración de herramientas informáticas que ayuden a mostrar y recuperar la información contenida un solo punto será de gran ayuda para dar cumplimiento a un proceso que requiere una comprensión absoluta del comportamiento y desempeño de un método analíticos durante todo su ciclo de vida.
Los alcances tecnológicos manifiestan avances de gestión que favorecen la mejora continua y el desempeño de las actividades de laboratorio.
La gestión y mantenimiento desde una perspectiva tecnológica se precisa de la siguiente manera:
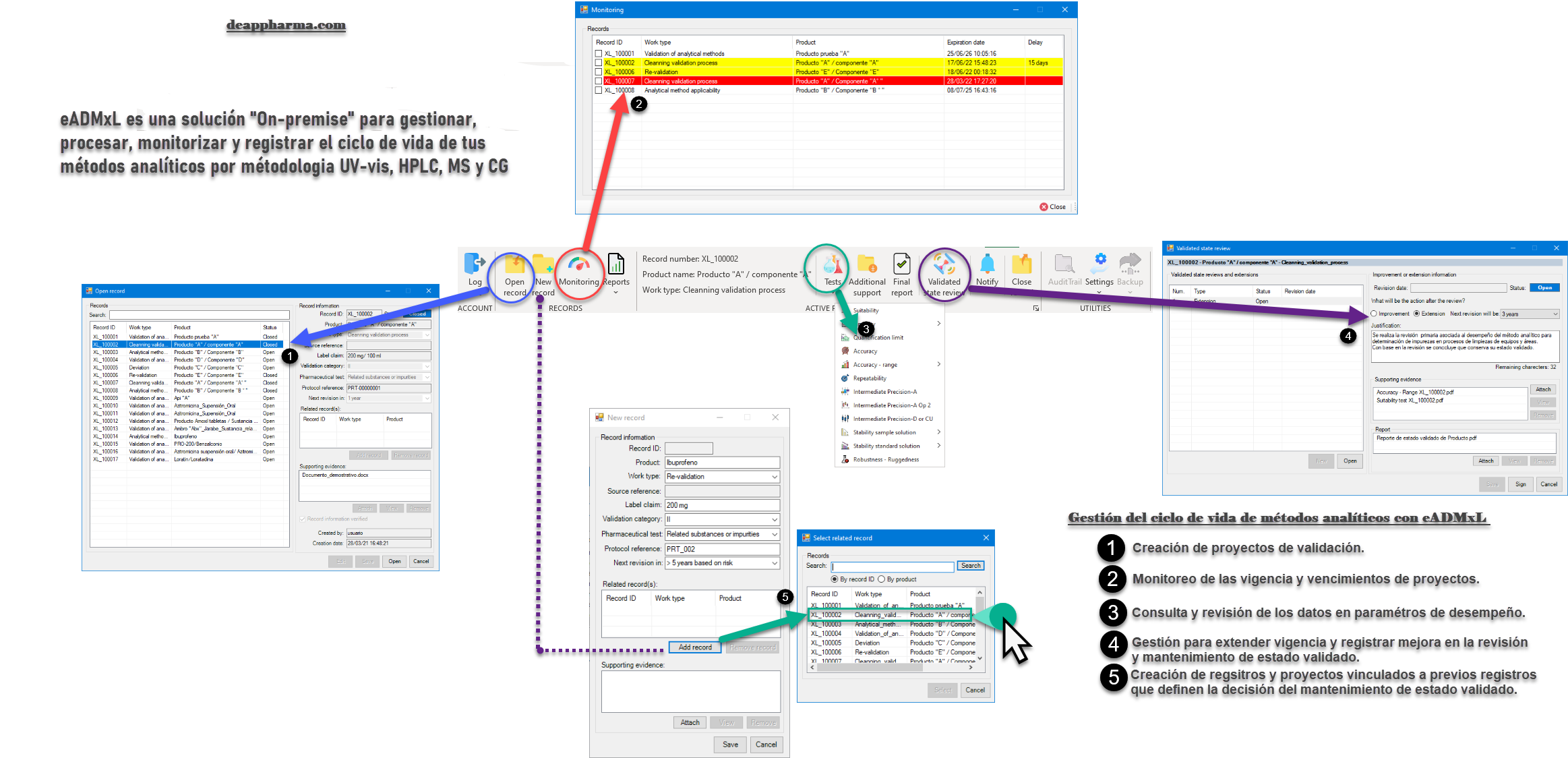
No olvides que, en caso de ser necesario, puedes contactarnos para mejorar el proceso de laboratorio y administrativo de tu organización a través software 100% confiable y seguro. Recuerda que en deappharma contamos con un software que te ayudara a controlar tu proceso de validación de métodos analíticos. Somos expertos en la materia, por lo que procesos complejos no representa un reto para nosotros, debido a que tenemos en mente los aspectos relevantes de cumplimiento.
Referencias
-
Analytical procedure development Q14 Draft version Endorsed on 24 March 2022
-
Validation of analytical procedures Q2(R2) Draft version Endorsed on 24 March 2022
¡Contáctanos! y conoce de que manera te vamos a ayudar.
¡Únete a nuestra comunidad en redes!
29, Jun 2021
Revisión de estado validado de método analíticos.
Uno de los varios procesos de control analítico dentro de la industria, es el seguimiento o mantenimiento del estado validado y/o estado de control de métodos analíticos. No es una tarea fácil!! ya que para realizar una correcta revisión, el entorno en el que gira el proceso de mantenimiento validado radica en el acceso a documentos y en la obtención de datos que se asocian directamente a la esencia del método, como son: primera validación y parámetros de adecuabilidad históricos.
Concepto:
El concepto de ciclo de vida, vincula el desarrollo de productos y procesos, por lo que se debe adquirir conocimiento para abordar la revisión.
Validación métodos y calidad de los medicamentos.
La validación efectiva del método contribuye significativamente a asegurar la calidad de los medicamentos. Lo básico, es comprender el principio de garantía de calidad. Este principio describe que se debe producir un medicamento que sea apto para el uso previsto.
Este principio incorpora el entendimiento de que existen las siguientes condiciones:
a) La calidad, la seguridad y la eficacia están diseñadas o integradas en el producto.
b) La calidad no puede garantizarse adecuadamente simplemente mediante el producto en proceso y terminado, inspección o prueba.
Un programa de validación exitoso depende de la información y el conocimiento del método, así como, de la comprensión del proceso de desarrollo analítico. Este conocimiento es la base para establecer un enfoque para el control del analítico, que da como resultado certidumbre en la calidad y atributos evaluados. Los científicos analíticos deben:
- Comprender las fuentes de variación (se puede evaluar durante la validación o con la revisión de los datos de validación).
- Detectar la presencia y el grado de variación (análisis de datos obtenidos en la validación y en el histórico de datos de liberación, sin embargo, esto no mide directamente el método).
- Controlar la variación de una manera acorde con el riesgo, que representa para el método que afecta los datos de liberación del producto.
Descripción del proceso analítico en tres etapas:
Etapa 1 – Diseño del proceso analítico: el proceso analítico se define durante este etapa, basada en el conocimiento adquirido a través de actividades de desarrollo.
Etapa 2 – Validación del proceso analítico: durante esta etapa, el diseño analítico se evalúa para determinar si método es capaz de producir resultados reproducibles.
Etapa 3 – Verificación continua del proceso analítico: la garantía continua se obtiene durante la rutina analítica, de tal manera, que se pueda verificar que el proceso permanece en un estado de control.
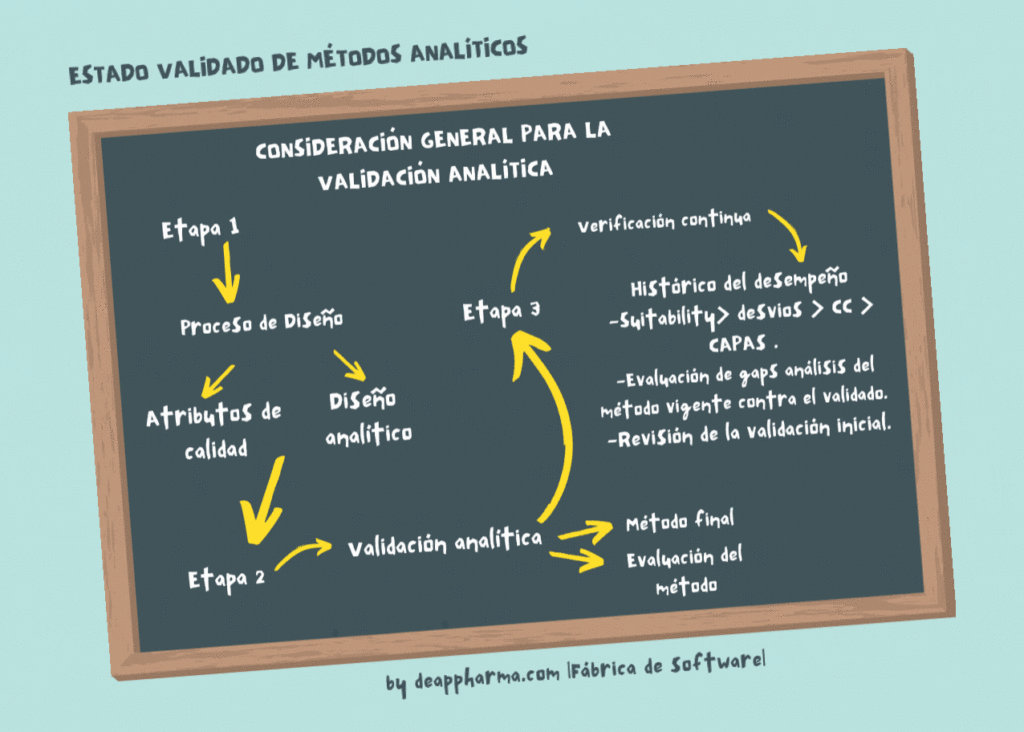
Los puntos relevantes de evaluación a la etapa 3, son:
- Requisitos de idoneidad históricos del sistema (platos teóricos, resolución, factor de coleo, factor de simetría, factor de capacidad y/o tiempos de retención de componentes).
- Parametrización y evaluación de las pruebas de desempeño acordes al método analítico.
- Revisión de los ajustes a las condiciones de operación indicadas en el procedimiento analítico vigente vs los descritos en la validación y/o transferencia inicial.
- Hallazgos o desviaciones, directamente relacionadas al desempeño del método analítico.
Si una evaluación basada en riesgos es utilizada como herramienta, se deben considerar otros factores. Los cuales pueden conducir a cambios en un procedimiento analítico y/o reemplazo con un nuevo método, se puede considerar los siguiente:
- Un ejercicio nuevo de transferencia;
- Una posible revalidación, sea parcial o total, o,un nuevo ejercicio de validación o,
- Un estudio de comparabilidad de métodos analíticos, es una opción factible.
En algunos casos, cambios en el fármaco o sustancias dentro del proceso de fabricación también deben ser evaluados como parte de la revisión de estado validado.
Recurso analítico «software».
Como se menciono anteriormente, la revisión de estado validado de un método analítico radica en que tan disponible y correcta fue la ejecución de este. Por lo que, la disposición de la información debe ser consistente. De otra manera será difícil concluir.
eADMxL el software de cumplimiento de deappharma.com , le ayudara con el control del proceso de estado validado mediante sus funciones especificas de «Revisión de estado validado» y «Monitoreo de vigencias».
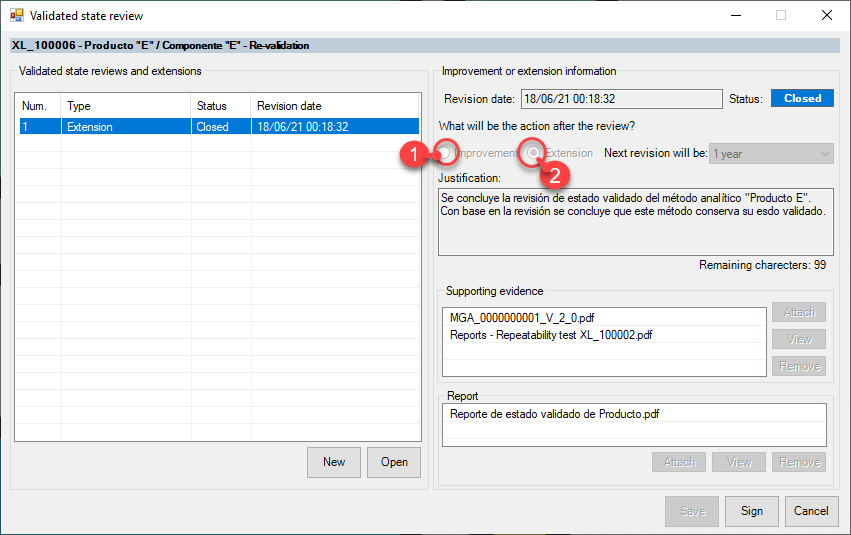
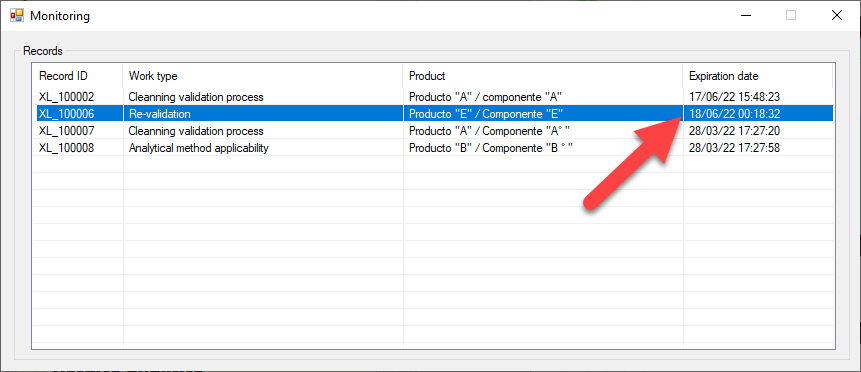
Algunas preguntas relevantes, que le ayudaran a definir el estado de control y/o mantenimiento, son:
- El método fue planteado y validado en concordancia con su categoría?
- La estadística paramétrica es integra?
- Los resultados de validación analítica son consistentes y claros?
- Si es un método compendial, los datos de pruebas de robustez tolerados están documentados?
- Los criterios de “Suitability” del sistema, se mantienen a través de los análisis?
- Las condiciones analíticas establecidas en el método final (preparación , condiciones de análisis, columna etc.) son las mismas que están descritas en el reporte de validación del método?
- Si hay cambios al método, se registran y se alinean a un control de cambio y se emite un CaPa al método y con esto evaluar el impacto a la validación?
- Qué tan recurrentes son las desviaciones o justificaciones asociadas al método analítico?
- Si la validación analítica sigue vigente, puedo hacer un revisión de estado validado antes de su fin expiración si surgen problemas?
No olvides que en caso de ser necesario, puedes contactarnos para mejorar el proceso de laboratorio y administrativo de tu organización a través software 100% confiable y seguro. Recuerda que en deappharma somos expertos en la materia, por lo que procesos complejos no representa un reto para nosotros, debido a que tenemos en mente los aspectos relevantes de cumplimiento.
Referencias.
- Analytical Procedures and Methods Validation for Drugs and Biologics Guidance for Industry
- Pharmaceutical quality system ICH Q10.
- Guidance for Industry Process Validation: General Principles and Practices.
¡Contáctanos! y conoce de que manera te vamos a ayudar.
¡Únete a nuestra comunidad en redes!