20, Jun 2024
GAMP 5 y la categorización de software
Cuando abordas el desafío para validar sistemas computarizados debes considerar como etapa previa la categorización del sistema computarizado. Como bien sabemos la normatividad regulatoria referente NOM 059 para medicamentos y la NOM 241 para dispositivos médicos. No definen las categorias existentes asociadas a los sistemas computarizados. Estas normas de manera general solo hacen mención bajo lo siguiente:
“ 9.13.4. El proceso de validación debe abarcar todas las fases relevantes del ciclo de vida de acuerdo a la categoría y arquitectura del sistema, para asegurar la exactitud, integridad y consistencia en el desempeño previsto de los Sistemas Computacionales”
Por este hecho, hay que hacer uso de guías internacionales tales cuales nos brindaran soporte para poder definir dicho requerimiento. Una de las principales guías para tomar como referencia este concepto, es la GAMP5. La cual establece de manera simplificada pero concisa la categorización de sistemas computarizados y con esto poder abordar el proyecto de validación aplicable.
Hay dos maneras de utilizar las categorías para definir alcances, siendo los siguientes:
- Evaluación de sistemas completos: a nivel de todo sistema, se puede definir la categoría del componente principal para ayudar con el enfoque de evaluación de proveedor. Combinando esta categorización con la evaluación de impacto sobre las buenas prácticas (BxP) que el sistema manifiesta. Esto con el fin de ayudar a decidir la extensión requerida para la evaluación del proveedor, según aplique.
- Evaluación de riesgo funcional: Una evaluación de riesgo funcional para la categorización puede ayudar a incrementar la objetividad en la evaluación del proceso. El mayor riesgo se deriva de una elevada combinación entre mayor complejidad y menor experiencia de usuario. Una compresión de la categoría del software puede contribuir para evaluar los siguientes riesgos: riesgos de fallas y/o defectos y/o riesgos donde la detectabilidad de fallos depende de una función del sistema.
GAMP 5 establece las siguientes categorías de software: (1) Software de infraestructura; (3) Componentes estándar de sistema; (4) Componentes configurables; (5) componentes y aplicaciones personalizadas. A continuación, abordaremos la descripción general para cada categoría.
Categoría 1- Infraestructura de software, herramientas, y servicio de IT
Descripción: Software en capas, software usado para administrar los ambientes de operación e infraestructura. Software y/o sistemas que dan soporte a sistemas computarizados dentro de las actividades del ciclo de vida. Ejemplo; motores de base de datos, sistemas operativos, lenguajes de programación, herramientas de supervisión de redes y rendimientos, herramientas de programación, software y/o sistemas como herramientas de soporte para procesos de IT.
Categoría 3- Componentes estándar de sistemas
Descripción: Software donde se pueden introducir y almacenar parámetros en el momento de ejecución. Pero, no se pueden configurar los componentes del sistema para adaptarlos a los procesos empresariales. Ejemplo; aplicaciones basadas en firmware y COTS los cuales son productos comerciales disponibles en el mercado (commercial-off-the-shelf o COTS) listos para usar, que se adaptan a posteriori a las necesidades de la organización compradora, en lugar de encargar soluciones personalizadas o a medida.
Categoría 4-Componentes configurables
Descripción: Software regularmente complejo, que puede ser configurado por el usuario para introducir requerimientos específicos para el proceso del negocio. El código de software no es alterado. Ejemplo; LIMS, SCADA, ERP, Monitoreo de pruebas clínicas, DCS, ARD de reporteo, CDS, EDMS, CRM, hojas de cálculo.
Categoría 5- Componentes y aplicaciones personalizadas
Descripción: Software diseñado y codificado a medida para adaptarse al proceso del negocio. Ejemplo; interfaces a la medida, aplicaciones de IT desarrolladas de manera interna o externa, aplicaciones desarrolladas de manera interna o externa para procesos de control, firmware personalizado, hojas de cálculo con macros.
Estamos convencidos de que la categorización de software depende de la evaluación de impacto y enfoque basado en el riesgo. Sin embargo, el alcance de cada sistema y su evaluación individual determinara el grado de detalle en la ejecución de prueba dentro de la validación.
¡Recuerda! que en deappharma tienes un aliado para realizar la validación de sistemas computarizados. ¡Con nuestro método obtendrás el cumplimiento requerido!
Nuestro servicio te da acceso a un beneficio totalmente gratis por un año. Este beneficio consiste en implementar eDocuSeed un software que centraliza todos tus libros de Excel y documentos en un solo punto. De esta manera les brindamos los atributos de integridad de datos, trazabilidad y mantenimiento necesarios para dar cumplimiento a las regulaciones más exigentes. Nuestra solución ya impacta de manera positiva a empresas del sector farmacéutico. Solicita tu demostración totalmente gratuita. ¡Estamos listos para ayudarte!
Contáctanos y solicita una demostración y asesoría gratuita ¡Quiero un asesoría y demostración!
¡Gracias por leernos!
- 0
- Por Team deappharma

26, Abr 2024
Revisión del diseño y trazabilidad en sistemas computarizados
Revisión de diseño
La revisión del diseño evalúa los entregables a través de requerimientos estándar, identificando eventos, y proponer las medidas correctivas necesarias. Esta revisión es sistemática y planeada, de tal manera que pueda ser desarrollada en todos los puntos del ciclo de vida del sistema. Esto es una de las partes importantes de la verificación del proceso.
La revisión del diseño deberá ser realizada por un equipo de especialistas apropiado. La revisión de desempeño individual deberá estar identificada. En algunos casos el uso de herramientas podría simplificar aspectos para la revisión de diseño; por ejemplo, análisis de código y herramientas de trazabilidad pueden proporcionar un enfoque eficiente de confianza.
El rigor de la revisión del proceso de diseño y la extensión de la documentación deben estar basadas en el riesgo, complejidad y novedad del sistema a evaluar.
Aspectos que deben ser considerados cuando se planea una revisión de diseño incluyen:
- El alcance y objetivo de la revisión.
- Que método y/o proceso deberá ser seguido.
- Quienes estarán involucrados, con roles y responsabilidades específicas.
- Cuáles serán los resultados.
Para productos estándar (GAMP categoría 3) la revisión de diseño en compañías reguladas típicamente no es requerida. Sin embargo, esto debe estar descrito en el riesgo definido.
Para sistemas basados en configuraciones de producto, gran parte de la revisión de diseño deberá ser realizada por el proveedor durante el desarrollo del producto. Esto deberá ser una parte de evaluación del proveedor. Las actividades de revisión de diseño deberán estar enfocadas en la configuración y las actividades de implementación.
Par aplicaciones personalizadas, las revisiones de diseño típicamente son conducidas a cada nivel del detalle de la especificación de diseño y alcance del producto.
Trazabilidad (Matriz de trazabilidad)
La trazabilidad establece relaciones entre dos o más productos en el proceso de desarrollo.
La trazabilidad asegura que:
- Los requerimientos han sido realizados y estos pueden ser trazados como parte de los elementos de diseño.
- Los requerimientos han sido verificados y que estos pueden ser trazados por medio de las actividades de prueba y verificación descritas en los requerimientos.
La exactitud en la trazabilidad puede proveer beneficios como:
- Permitir una mayor eficacia en la administración de riesgos.
- Juzgar impactos potenciales a cambios propuestos.
- Facilitación para la estructuración del análisis de riesgos por cambios propuestos.
- Identificar alcance de evaluación de cambios.
- Permitir mayor exactitud y rapidez durante una inspección de auditoría.
¡Recuerda! que en deappharma tienes un aliado para realizar la validación de sistemas computarizados. ¡Con nuestro método obtendrás el cumplimiento requerido!
Con nuestro servicio obtendrás un beneficio totalmente gratis por un año. Este beneficio consiste en implementar eDocuSeed un software que centraliza todos tus libros de Excel en un solo punto. Para brindarles los atributos de integridad de datos, trazabilidad y mantenimiento necesarios para dar cumplimiento a las regulaciones más exigentes. Nuestra solución ya impacta de manera positiva a empresas del sector farmacéutico, dispositivos médicos y distribuidoras de medicamentos. Solicita tu demostración totalmente gratuita. ¡Estamos listos para ayudarte!
Contáctanos y solicita una demostración y asesoría gratuita ¡Quiero un asesoría y demostración!
¡Gracias por leernos!
23, Ene 2024
Requerimientos generales para la validación de sistemas computarizados
Cuando hablamos de sistemas computarizados debemos tener en cuenta ciertos principios de validación que pueden repercutir la calidad de los resultados, el control de documentos y el almacenamiento de datos. El objetivo de la validación es garantizar la confianza en los datos de laboratorio capturados, procesados, notificados o almacenados por los sistemas informatizados. Un sistema validado garantiza la exactitud de los resultados y reduce los riesgos para la integridad de los datos. A continuación, abordaremos los requerimientos generales establecidos por la OMCL (Official Medicines Control Laboratories) de la unión europea (EDQM) para la validación de sistemas computarizados.
Inventario
Se debe mantener un inventario o una lista equivalente debe estar disponible. La información mínima que debe incluir el inventario de sistemas computarizados, es:
-Identificación y versión
-Propósito
-Estatus de la validación
-Tipo de almacenamiento
-Persona de contacto
Validación
Antes de su uso rutinario, el sistema informatizado deberá ser validado.
El objetivo de la validación es confirmar que las especificaciones del sistema informatizado se ajustan a las necesidades del usuario y los usos previstos mediante el examen y la aportación de pruebas objetivas y que los requisitos particulares pueden cumplirse de forma coherente.
El alcance de la validación dependerá de la complejidad y del uso previsto del sistema informatizado que se valide.
El esfuerzo de validación puede escalarse y adaptarse al tipo de sistema justificado por una evaluación de riesgos documentada.
Registro de problemas
Debe mantenerse un registro de los problemas identificados por los usuarios y de las medidas adoptadas.
Control de cambios
En caso de cambios en el sistema informático, incluidas las actualizaciones de versión, lo ideal sería en un entorno de prueba, tras lo cual deberá restablecerse el estado de validación.
Si es necesaria una revalidación, debe realizarse no sólo para validar el cambio individual, sino
también para determinar el alcance y el impacto de ese cambio en todo el sistema informatizado.
El alcance de la revalidación dependerá de la evaluación del cambio o cambios, que deberá estar
documentada. Un posible enfoque podría ser el uso de cuadernos de bitácora como se hace para los equipos, y/o la utilización de un procedimiento documentado de control de cambios.
Revisión periódica
El OMCL debe adoptar una política de comprobación periódica del sistema informatizado para evitar cualquier error y garantizar el mantenimiento del estado de validación. La frecuencia de las revisiones deberá definirse en función de los riesgos. Los sistemas informatizados deberán estar cubiertos por la estrategia de auditoría interna.
Seguridad y condiciones ambientales
Los sistemas informatizados deben estar protegidos contra cualquier intrusión que pueda modificar los datos y afectar a los resultados finales.
Las salas de servidores deben tener acceso restringido y contar con las condiciones necesarias para garantizar el correcto funcionamiento de los equipos (control de temperatura, medidas contra incendios, Sistema de Alimentación Ininterrumpida, etc.).
El acceso a los sistemas debe estar restringido al personal autorizado, mediante cuentas personalizadas y contraseña o método de identificación equivalente. Debe evitarse el uso de cuentas compartidas y genéricas para garantizar que las acciones documentadas en los sistemas informáticos puedan atribuirse a una única persona. Cuando no se disponga de cuentas personales o éstas no sean viables, deberán utilizarse combinaciones de registros en papel y electrónicos para rastrear las acciones del personal responsable.
Sólo la(s) persona(s) responsable(s) o el personal informático designado deben tener derechos administrativos para implementar cualquier actualización y/o instalación de sistemas informáticos, cambiar ajustes críticos del sistema (por ejemplo, registro de auditoría, hora/fecha) y gestionar los permisos de otros usuarios. Todas las tareas rutinarias, como las de análisis, deben basarse en una cuenta de usuario y una contraseña que no tengan derechos administrativos.
Los derechos administrativos deben documentarse y concederse únicamente al personal con funciones de mantenimiento del sistema (por ejemplo, informáticos) que sean totalmente independientes del personal responsable del contenido de los registros (por ejemplo, analistas de laboratorio, dirección del laboratorio).
Cuando no sea posible asignar de seguridad independientes, deberán utilizarse otras estrategias de control para reducir los riesgos para la integridad de los datos.
Los ordenadores deben bloquearse después de su uso y no debe permitirse a los usuarios cambiar los ajustes de fecha y hora.
El hardware utilizado debe cumplir los requisitos técnicos para que el trabajo se pueda realizar. Dichos requisitos incluyen, por ejemplo, los requisitos mínimos del sistema indicados por el fabricante del sistema. Estos requisitos deben estar predefinidos en función del uso previsto.
Los componentes de hardware deben ser instalados por personal cualificado (por ejemplo, personal de la Unidad de Tecnologías de la Información (TI), un técnico del fabricante del equipo, etc.).
Registro de auditoría
El sistema informático debe mantener un registro de todas las acciones críticas que se produzcan, por ejemplo, quién ha accedido a él y cuándo, cualquier supresión o modificación de datos, etc. Si un sistema informatizado no registra automáticamente una pista de auditoría, el OMCL deberá mantener un registro alternativo.
No se permitirá a los usuarios modificar o desactivar las pistas de auditoría o los medios alternativos de proporcionar trazabilidad de las acciones de los usuarios.
En todos los sistemas informatizados nuevos deberá tenerse en cuenta la necesidad de implantar una función de pista de auditoría adecuada.
Cuando un sistema informatizado existente carezca de pistas de auditoría generadas por ordenador, el personal utilizará medios alternativos como el uso controlado por procedimientos de libros de registro, control de cambios, control de versiones de registros u otras combinaciones para cumplir el el requisito de trazabilidad para documentar el qué, quién, cuándo y por qué de una acción.
Firmas electrónicas
Si se utilizan firmas electrónicas, una declaración sobre la equivalencia de la firma electrónica a la firma manuscrita o declaración legal similar debe existir dentro del sistema de gestión de la calidad.
Archivo de versiones sustituidas de sistemas informáticos
Las versiones sustituidas de los programas informáticos deben archivarse de forma recuperable si es necesario para acceder a datos históricos, de acuerdo con la directriz de la OMCL «Gestión de documentos y registros».
Para el caso de los programas informáticos comerciales, la obligación de archivar las versiones anteriores puede estar sujeta al contrato con el proveedor.
Formación
Se garantizará la correcta utilización y validación del sistema informático. Esto puede hacerse mediante una formación adecuada y documentada o mediante información detallada en los procedimientos pertinentes o información contextual en el programa informático.
La formación se impartirá antes del primer uso y después de cada cambio importante en el software (por ejemplo, actualización de la versión). Las personas responsables de la validación recibirán formación sobre el proceso de validación.
¡Recuerda! que en deappharma tienes un aliado para realizar la validación de sistemas computarizados. Con nuestro método obtendrás el cumplimiento requerido.
Con nuestro servicio obtendrás un beneficio totalmente gratis por un año. Este beneficio consiste en implementar eDocuSeed un software que centraliza todos tus libros de Excel en un solo punto. Para brindarles los atributos de integridad de datos, trazabilidad y mantenimiento necesarios para dar cumplimiento a las regulaciones más exigentes. Nuestra solución ya impacta de manera positiva a empresas del sector farmacéutico, dispositivos médicos y distribuidoras de medicamentos. Solicita tu demostración totalmente gratuita. ¡Estamos listos para ayudarte!
Contáctanos y solicita una demostración y asesoría gratuita ¡Quiero un asesoría y demostración!
¡Gracias por leernos!
31, Oct 2023
Validación de hojas de cálculo (Instalación, seguridad y buenas prácticas)
En industria regulada es de vital importancia el cumplimiento a lo descrito en las normas aplicables: NOM-059, NOM-241 etc. Recordemos que el cumplimiento a los requerimientos establecidos en estos documentos es de carácter obligatorio. Es una realidad que el hacer cumplir los requerimientos en muchos casos se torna una tarea compleja por la dimensión de interpretación que le puedas dar a cada requerimiento regulatorio establecido. Sin embargo, es importante mencionar que existen una serie de documentos, guías y referencias de otras entidades regulatorias que dan apoyo y complementan de manera correcta la interpretación para aquellos requerimientos en los cuales no sea claro el entregable.
Cuando hablamos de validación de sistemas computarizados debemos tener presente que los puntos normativos no solo se refieren a validar: ERP, SAP, softwares comerciales, softwares personalizados o softwares estructurados y diseñados a las medidas, por mencionar algunos. Recordemos que una hoja de Excel normativamente se considera un sistema computarizado. Tales cuales, en muchas ocasiones estos archivos son categorizados con en nivel máximo de complejidad y criticidad que impactan los procesos de las buenas prácticas BxP (GxP).
Esta serie de artículos tendrá como finalidad explicarte el enfoque de validación de hojas de cálculo establecido en el Anexo 1 de la Directriz Europea.
Durante nuestra experiencia hemos conocido diferentes magnitudes de riesgos asociados al control, gestión y mantenimiento de hojas de cálculo. Hemos conocido empresas que no cuentan con área de validación o empresas que por la rutina diaria no disponen del tiempo para ejecutar la acción. De cualquier manera, esto no los deja exentos de cumplir la normatividad.
Todos sabemos que Excel es una herramienta altamente versátil y dinámica utilizada diariamente. Sin embargo, esta versatilidad le impregna un alto riesgo por el grado de personalización que estas pueden llegar a tener en su diseño y función y que impactan en la toma de decisiones.
El Anexo 1 de la Directriz Europea para la validación de hojas de cálculo «Validación de sistemas informatizados», debe utilizarse a la hora de planificar, realizar y documentar la validación.
Este anexo presenta un ejemplo de validación de hojas de cálculo Excel, que debe utilizarse en combinación con los requisitos y recomendaciones generales que figuran en el documento referente y/o lo establecido en la regulación local aplicable.
INSTALACIÓN Y SEGURIDAD
Para garantizar que sólo se utiliza la última versión validada de la hoja de cálculo y mantener el estado validado de la hoja de cálculo, todas las hojas de cálculo Excel validadas deben almacenarse con derechos de acceso de sólo lectura para los usuarios finales (por ejemplo, en un recurso compartido de red protegido o algo con mucha mayor robustez en seguridad un software de control y mantenimiento como lo es DocuSeed). Sólo las personas responsables deben tener acceso de escritura a la red compartida.
Los usuarios finales no deben tener derecho a modificar una hoja de cálculo validada, añadir una hoja de cálculo no validada al recurso compartido ni guardar datos en él. Los usuarios finales sólo deben tener derecho a rellenar las celdas (permitidas) e imprimir los datos o guardar una copia en un repositorio de datos en caso necesario.
La instalación deberá documentarse, por ejemplo, en el archivo de validación, en un libro de registro del sistema o en un formulario de control de calidad. Se documentará el nombre de la hoja de cálculo, la identificación única, la localización y la persona responsable de la hoja de cálculo según aplique. Los registros también incluirán la verificación, la verificación periódica y otras cuestiones como las actualizaciones o cualquier problema encontrado. La verificación se completará tras instalación y se registrará.
BUENAS PRÁCTICAS
Al configurar una nueva hoja de cálculo, seguir las siguientes buenas prácticas reducirá el riesgo de modificaciones accidentales de la plantilla e introducción de datos erróneos:
– Todas las celdas de cálculo deberán estar bloqueadas (Formato de celdas > Protección > Bloqueadas) para proteger las celdas que contengan cálculos contra modificaciones involuntarias, excepto las utilizadas para la introducción de datos.
-Las celdas utilizadas para la introducción de datos pueden identificarse mediante un color específico.
– Las reglas de validación de datos (pestaña Datos > Validación de datos) pueden aplicarse a las celdas de entrada de datos para evitar la introducción de valores aberrantes (siempre y cuando exista un requerimiento de especificación previamente establecido). Los mensajes de entrada y los mensajes de alerta de error para informar al usuario final del tipo de datos esperado y del rango aceptable.
Las celdas utilizadas para presentar los resultados de los cálculos (salida) pueden identificarse mediante un color específico. Cuando los resultados se comprueban en función de criterios de aceptación, se recomienda utilizar el formato condicional (pestaña Inicio > Formato condicional) para resaltar los resultados fuera de especificación.
-El nombre del operador responsable de la introducción de los datos, así como la fecha y la hora de introducción de los datos deben registrarse en las celdas de entrada específicas o la hoja de cálculo se imprime, firma y fecha después del cálculo.
– La ruta del archivo, el nombre del archivo de la hoja de cálculo y el número de versión de MS Excel® pueden mostrarse en el área de impresión de la hoja de cálculo. Las funciones de Excel ‘=CELL(«filename»)’ ‘=INFO(«RELEASE»)’ pueden utilizarse para mostrar la ruta, el nombre de archivo, la hoja activa y el número de versión de MS Excel® en uso.
-Se recomienda proteger con contraseña todas las celdas que contengan cálculos (pestaña Revisar >Proteger hoja), con sólo las opciones por defecto marcadas. Se puede utilizar la misma contraseña para todas hojas y puede documentarse en el archivo de validación. La contraseña de protección de la hoja no debe comunicarse a los usuarios finales.
-Después de proteger cada hoja, la estructura del libro de trabajo también debe protegerse con contraseña (pestaña Revisar > Proteger libro). Se puede utilizar la misma contraseña que para la protección de las hojas.
Como lo podemos percibir la etapa de validación de hojas de cálculo esencialmente refiere a la estructuración documental del diseño y funcionamiento para el propósito requerido. En esta etapa no se establece como alcance regulatorio el mantenimiento de estos archivos con propiedades ALCOA++. El tema de fases de validación y mantenimiento de estos archivos los abordaremos en otro artículo.
Te invitamos a conocer una solución confiable que te ayudara a brindar atributos ALCOA++ a tus hojas de cálculo validadas. En deappharma hemos desarrollado, eDocuSeed un software que centraliza todos tus libros de Excel en un solo puntos brindándoles los atributos de integridad de datos necesarios para dar cumplimiento a las regulaciones más exigentes. Nuestra solución ya impacta de manera positiva a empresas del sector farmacéutico, dispositivos médicos y distribuidoras de medicamentos. Solicita tu demostración totalmente gratuita. ¡Estamos listos para ayudarte!
Contáctanos en https://deappharma.com/contacto/ y solicita una demostración y asesoría gratuita.
¡Gracias por leernos!
3, Jul 2023
Hojas de cálculo; categorización conforme a GAMP 5
Las hojas de cálculo pueden ser tan complejas o simples según la necesidad de la actividad a realizar. Sin embargo, al momento de evaluar una hoja de cálculo el proceso es mucho más sistemático de lo que se cree. Te invitamos a leer nuestro siguiente articulo y conoce algunos detalles relevantes que te ayudaran a categorizar tu hoja de cálculo.
Enfoque basado en el riesgo
Una aplicación de hoja de cálculo puede variar significativamente en cuanto a riesgo y complejidad. No obstante, en todos los casos es necesario cumplir los siguientes requisitos:
- Evaluación del riesgo y medidas de control del riesgo apropiadas para gestionar el riesgo identificado.
- Especificación y verificación apropiadas para demostrar que la aplicación funciona según lo previsto.
La estrategia de especificación y verificación de la aplicación que se está construyendo debe basarse en:
- El impacto del sistema en la seguridad del paciente, la calidad del producto y la integridad de datos mediante la evaluación del riesgo.
- Complejidad y novedad del sistema (arquitectura y categorización de los componentes del sistema).
- La seguridad apropiada para mitigar el riesgo de cambios no autorizados en los datos o la aplicación.
Es importante mencionar que el proceso de validación de sistemas computarizados en los cuales están incluidas las hojas de cálculo deben estar regidas por políticas y procedimientos de la empresa para definir y lograr mantener la conformidad y la idoneidad para el uso previsto de la aplicación por el usuario final.
Uso de categorías GAMP 5
La categorización de hojas de cálculo se debe establecer con base en la guía GAMP 5. La guía nos proporciona lineamiento y algunos ejemplos generales que nos ayudan a comprender el rumbo a tomar para realizar el proceso de validación de este tipo de archivos.
GAMP 5 establece y define 4 categorías de clasificación siendo las siguientes; 1, 3, 4 y 5. A continuación se describen criterios y detalles con base en esta Guía.
La herramienta sobre la que se construye la aplicación, como el paquete de hoja de cálculo, debe considerarse como categoría 1.
Las categorías para hojas de cálculo y otras aplicaciones de usuario final deben considerarse como un continuo que abarca las categorías 3, 4 y 5. La asignación de una categoría depende de la complejidad y personalización de la hoja de cálculo o aplicación. No obstante, hay que tener en cuenta que una hoja de cálculo que se limita a utilizar la capacidad de esta para edición, tabulación y no realiza ningún cálculo debe considerarse un documento. El uso previsto de los datos ensayados debe tenerse en cuenta a la hora de determinar los requisitos de verificación y el control que debe establecerse. Una simple hoja de cálculo (categoría 3) puede presentar un riesgo BxP (o GxP) elevado en función del uso que se haga de los datos.
Una hoja de cálculo que simplemente utiliza funciones nativas para realizar cálculos en lugar de una calculadora de mano suele ser de categoría 3. Por ejemplo, un analista de laboratorio puede crear una hoja de cálculo única para realizar un cálculo relacionado con una investigación derivado de un fuera de especificación. Cuando se utiliza la función aritmética de las hojas de cálculo, el cálculo debe explicarse detalladamente. Los datos deben explicarse de la misma manera que en un documento de texto. Esto debe incluir la verificación de que el cálculo se ha realizado correctamente y que los datos analizados son los correctos. Esta verificación podría documentarse fácilmente haciendo que otro analista o un supervisor examinara la hoja de cálculo y la aprobara. No se requiere ninguna otra verificación, ya que no es necesario cuestionar la exactitud del cálculo.
Cuando se desarrollen hojas de cálculo como templetes, podrían ser de categoría 3 a 5, dependiendo de la complejidad. Para el propósito de la aplicación de usuario, la definición de la categoría 4 se altera ya que esta se centra en la complejidad frente a la configurabilidad.
Por ejemplo:
- Un analista utiliza un templete en el laboratorio para realizar un cálculo rutinario de medias y desviación estándar de resultados experimentales. Se trata de una operación aritmética directa sin configuración, por lo que el templete es de categoría 3.
- Un templete de hoja de cálculo requiere que el usuario introduzca la dureza de la tableta, por lo que la aplicación se ramifica automáticamente a diferentes celdas (customs) para utilizar el cálculo específico de la dureza basado en esta entrada. Una operación tan simple haría que la hoja fuera de categoría 4, ya que tiene alguna operación booleana (es decir, celdas vinculantes o condicionales lógicas a otras celdas para procesar datos de entrada) simple basada en la entrada del usuario.
- Una aplicación de hoja de cálculo que emplee macros personalizadas o una lógica anidada sofisticada para obtener resultados o una función de búsqueda debería tratarse como categoría 5.
En la siguiente ilustración podrás observar el alcance generar por aplicación y la relación que estas tienen respecto a sus diferentes categorías.
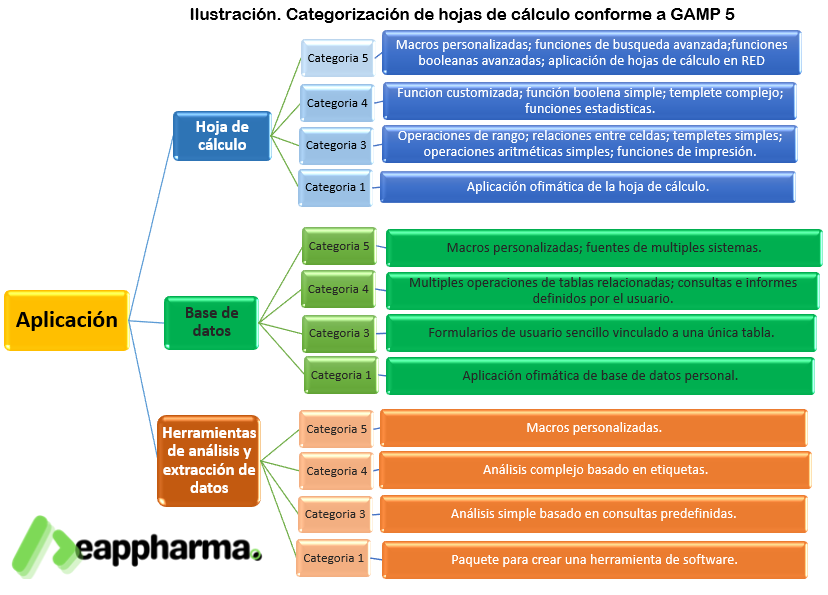
¿Existen herramientas en el mercado que mejoren los procesos de control y mantenimiento de libros de Excel?
Como bien sabemos el desafío más grande al que se ha puesto a prueba el sector farmacéutico es el relacionado con la integridad de datos. Derivado de la gran cantidad de datos emitidos día con día en el que se ven impactados documentos y libros de Excel.
Recordemos que la validación de hojas de cálculo es el primer paso. No obstante, muchos libros de Excel que contienen hojas de cálculo utilizados en el sector farmacéutico no cuentan con evidencia de validación lo cual es un riesgo que podrá derivar en un hallazgo critico ante una inspección regulatoria. El segundo paso y el más complejo es adherir las configuraciones necesarias que eleven la integridad de datos y favorezcan la gestión y el mantenimiento de estas con base en GAMP 5 y CFR21.
En la actualidad el trabajo realizado y el menos sugerible es la configuración de control de seguridad y trazabilidad sobre usos de hojas de cálculo con el uso de VBA lo cual es meramente simbólico que a mediano y largo plazo será poco sostenible en términos de cumplimiento e integridad de datos.
En deappharma tenemos la solución para mitigar tu riesgo por el uso de hojas de cálculo no validadas y con deficiencias en los atributos de control de integridad de datos. Hemos logrado automatizar el proceso y, con esto ahorres tiempo, dinero y esfuerzo para dar el cumplimiento regulatorio requerido. Nuestros desarrollos están estructurados bajo un sistema de gestión de calidad trazable y auditable y, tales cuales cuentan con el soporte de validación-calificación que garantizan la precisión del alcance del diseño establecido.
Estamos convencidos de que eDocuSeed te ayudara a mitigar el uso de hojas de cálculo y dar ese paso de cumplimiento que necesitas. Conoce sus beneficios y alcance general aquí: ¡Da clic aquí!
“No vivas con el riesgo ¡Mitígalo!” para eso nosotros de ayudamos.
Visita nuestro sitio www.deappharma.com y solicita una demostración y asesoría gratuita.
¡Contáctanos y únete a nuestra comunidad en redes!
6, Mar 2023
Importancia de la integridad de datos en procesos de validación de métodos analíticos
La integridad de datos es un aspecto crucial en el proceso de validación de métodos analíticos en la industria farmacéutica. La validación de métodos analíticos es un proceso que asegura que un método analítico específico es adecuado para su uso previsto y que proporciona resultados precisos y confiables.
La integridad de los datos es esencial en la validación de métodos analíticos porque los datos generados durante el proceso de validación se utilizan para establecer la precisión, la exactitud, la linealidad, la especificidad y la robustez del método analítico. La falta de integridad de los datos puede comprometer la precisión y confiabilidad de los resultados y poner en riesgo la seguridad y eficacia de los productos farmacéuticos.
Además, las autoridades reguladoras exigen que los datos utilizados en la validación de métodos analíticos sean precisos, confiables y completos. La falta de integridad de los datos puede poner en duda la validez de los resultados de la validación y poner en riesgo la aprobación de los productos.
Por lo tanto, la integridad de los datos es fundamental en el proceso de validación de métodos analíticos y se deben implementar controles rigurosos para garantizar que los datos generados durante el proceso de validación sean precisos, confiables y completos. Esto incluye la capacitación del personal, la implementación de controles de acceso y seguridad, la validación de sistemas, la verificación de datos y la revisión regular de la documentación y registros.
¿Cuales son los 5 hallazgos de integridad de datos en Hojas de cálculo en el sector farmacéutico?
A continuación de algunos hallazgos comunes de integridad de datos que se han observado en hojas de cálculo en sectores regulados, incluido el farmacéutico:
- Errores de entrada de datos: Los errores de entrada de datos son comunes en las hojas de cálculo y pueden deberse a errores humanos al ingresar información. Estos errores pueden incluir errores tipográficos, errores de transposición o errores de formato.
- Fórmulas incorrectas: Las fórmulas incorrectas pueden llevar a resultados inexactos y pueden deberse a una variedad de factores, como errores humanos, cambios en los datos de origen y copiar y pegar incorrectos.
- Control de cambios insuficiente: La falta de control de cambios puede hacer que sea difícil realizar un seguimiento de los cambios realizados en una hoja de cálculo y puede dificultar la identificación de errores o fraudes.
- Falta de validación de datos: La falta de validación de datos puede hacer que los datos sean inexactos o incompletos. Esto puede incluir la falta de validación de datos de entrada o la falta de validación de datos calculados por fórmulas.
- Falta de documentación: La falta de documentación puede dificultar la comprensión de cómo se creó una hoja de cálculo, lo que puede hacer que sea difícil identificar errores o fraudes. Es importante mantener una documentación completa y actualizada de las hojas de cálculo para garantizar la integridad de los datos.
Ahora que conoces la importancia de la integridad de datos en el proceso de validación de métodos analíticos y los hallazgos recurrentes en la industria vamos con el siguiente paso ¡Mejorar el proceso! ¿Quieres saber como?
¡DA CLIC AQUI!

¡Contáctanos! y conoce de que manera te vamos a ayudar.
¡Únete a nuestra comunidad en redes!
5, Abr 2022
ICH Q14 desarrollo de métodos analíticos y el impacto sobre ICH Q2 validación de métodos analíticos
Uno de los procesos GxP relevantes dentro de la industria es el proceso de validación de método analíticos. La importancia esencial es su vinculación directa con el método analítico utilizado de rutina (Liberaciones, Estabilidades). Con la actualización “propuesta” por parte de ICH, en lo referente a los procesos analíticos de desarrollo y validación se busca que estos tengan una relación estrecha en el que el beneficio principal es la calidad por diseño y mantenimiento del estado validado.
Como bien sabemos, el objetivo de la validación de procedimientos analíticos es demostrar que el procedimiento es adecuado para el fin previsto. Sin perder de vista que los datos y resultados deben ser suficientemente confiables para la toma de decisiones.
Un estudio de validación está diseñado para proporcionar pruebas suficientes de que el procedimiento analítico cumple sus objetivos. Estos objetivos se describen con un conjunto adecuado de características de rendimiento correspondientes, que pueden variar en función de uso previsto del procedimiento analítico y la tecnología especifica seleccionada.
Con la reciente propuesta de la guía ICHQ14 se busca incluir, describir y robustecer el proceso de laboratorio por la estrecha relación que existe entre el desarrollo de métodos y la validación de estos. Con esto podremos visualizar términos como son: estrategia de validación y control, ciclo de vida y mantenimiento como parte esencial de esta. Sin perder de vista la inclusión de metodologías existentes, tales cuales no eran claramente abordadas.
La propuesta ICH Q14 atribuye que dicho proceso y los conocimientos arrojados por este, se controlen de tal manera que se pueda obtener la información sobre la finalidad prevista del procedimiento analítico, y el rendimiento de las características y criterios asociados que deben ser validados.
La siguiente figura muestra cómo se puede generar el conocimiento durante el desarrollo de un procedimiento analítico propuesto en la ICHQ14 y con esto ayudar al diseño de un estudio de validación.
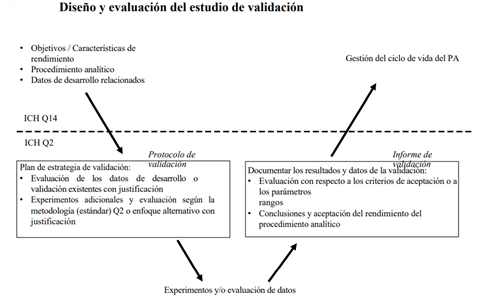
Validación durante el ciclo de vida de un procedimiento analítico
Durante el ciclo de vida de un procedimiento analítico pueden ser necesarios cambios. En tales casos, los cambios parciales pueden necesitar una revalidación completa. La ciencia y los principios basados en el riesgo pueden utilizarse para justificar sin una determinada característica de rendimiento necesita o no revalidación. El alcance de la revalidación depende de las características de rendimiento analítico afectadas por el cambio.
Consideraciones generales para el desarrollo de procedimientos analíticos y la gestión del ciclo de vida
El objetivo del desarrollo es obtener un procedimiento analítico adecuado para su propósito: medir un atributo o atributos del material analizado con la especificidad/selectividad, exactitud y/o precisión en el intervalo notificable. En esta sección se describen los enfoques mínimo y mejorado del desarrollo de procedimientos analíticos En esta sección se describen los enfoques mínimo y mejorado para el desarrollo de procedimientos analíticos. Aunque el enfoque mínimo sigue siendo aceptable, algunos o todos los elementos del enfoque mejorado de este enfoque mejorado para apoyar el desarrollo y la gestión del ciclo de vida de los procedimientos analíticos.
En ciertos casos, un procedimiento analítico establecido puede aplicarse a múltiples productos con poca o ninguna modificación de las condiciones de medición. Para una nueva aplicación de dichos procedimientos analíticos de plataforma plataforma, el desarrollo posterior puede abreviarse y algunas pruebas de validación puede omitirse sobre la base de una justificación científica y de riesgo. Los detalles de las características de rendimiento consideradas para la validación de procedimientos analíticos se describen en la ICH Q2.
En general, los datos obtenidos durante los estudios de desarrollo (por ejemplo, los datos de robustez de un diseño de de experimentos (estudio DoE)) pueden utilizarse como datos de validación para las características de rendimiento del procedimiento analítico características de rendimiento del procedimiento analítico y no es necesario repetirlos.
Enfoques mínimos frente a enfoques mejorados para el desarrollo de procedimientos analíticos
Enfoque mínimo.
El desarrollo de procedimientos analíticos debe incluir los siguientes elementos, según corresponda:
- Identificación de los atributos de la sustancia o el producto farmacéutico que deben ser analizados por el procedimiento analítico.
- Selección de una tecnología de procedimiento analítico apropiada y de los instrumentos o aparatos adecuados.
- Llevar a cabo estudios de desarrollo apropiados para evaluar el rendimiento del procedimiento analítico
- características de rendimiento del procedimiento analítico, como la especificidad, la exactitud y la precisión en el intervalo notificable (incluyendo el modelo de calibración, los límites en los extremos del rango inferior y/o superior) y la solidez.
- Definir una descripción adecuada del procedimiento analítico que incluya la estrategia de control estrategia de control del procedimiento analítico (por ejemplo, ajustes de los parámetros y adecuación del sistema).
Enfoque mejorado
El enfoque mejorado ofrece una forma sistemática de desarrollar y perfeccionar el conocimiento de un procedimiento analítico. Un enfoque mejorado debe incluir uno o más de los siguientes elementos, además de los ya descritos para el enfoque mínimo:
- Una evaluación de las propiedades de la muestra y de la variabilidad esperada de la muestra basada en conocimiento del proceso de fabricación.
- Definición del perfil analítico objetivo.
- Realización de una evaluación de riesgos y evaluación de los conocimientos previos para identificar los parámetros del procedimiento analítico que pueden afectar al rendimiento del procedimiento.
- Realización de experimentos uni o multivariados para explorar los rangos y las interacciones entre parámetros identificados del procedimiento analítico.
- Definir una estrategia de control del procedimiento analítico basada en una mejor comprensión del procedimiento incluyendo los puntos de ajuste y/o rangos apropiados para los parámetros relevantes del procedimiento analítico garantizando el cumplimiento de los criterios de rendimiento.
Definir un plan de gestión de cambios del ciclo de vida con definiciones claras y categorías de información de condiciones establecidas, rangos aceptables probados o regiones de diseño operacional del método según proceda. La aplicación de elementos del enfoque mejorado al desarrollo puede conducir a procedimientos analíticos más sólidos, a una mejor comprensión del impacto de los parámetros del procedimiento analítico y a una mayor flexibilidad para la gestión del ciclo de vida, como rangos operativos más amplios, un conjunto más apropiado de condiciones establecidas y categorías de información asociadas para los cambios.
El enfoque mejorado ofrece potencialmente varias ventajas, entre ellas
- Comprensión de aquellos atributos del procedimiento analítico tales cuales son esenciales para el rendimiento del procedimiento.
- Mejorar el control de los procedimientos analíticos para conseguir un funcionamiento más fiable.
- Permitir medidas preventivas y facilitar la mejora continua mediante el uso de más conocimiento de los procedimientos analíticos.
- Reducir la cantidad de esfuerzo a lo largo del ciclo de vida del procedimiento analítico.
El ciclo de vida del procedimiento analítico
La siguiente figura muestra los elementos del ciclo de vida del procedimiento analítico.
El desarrollo de procedimientos analíticos y los enfoques de gestión de cambios se describen en esta directriz, mientras que la validación de los procedimientos analíticos se describe en la ICH Q2. Dependiendo del uso previsto del procedimiento analítico y del enfoque de desarrollo adoptado, el orden y el alcance de cada elemento pueden variar, y varios elementos pueden darse simultáneamente.

Estrategia de control del procedimiento analítico
Una estrategia de control de procedimientos analíticos debe garantizar que el procedimiento analítico funcione como esperado durante su uso rutinario a lo largo de su ciclo de vida y consiste en un conjunto de controles, derivados de la comprensión actual del procedimiento analítico, incluidos los datos de desarrollo, la evaluación de riesgos y la robustez. El conocimiento previo también podría utilizarse para desarrollar la estrategia de control del procedimiento analítico.
La estrategia de control del procedimiento analítico debe definirse antes de la validación (ICH Q2) y debe confirmarse una vez finalizada la validación.
La estrategia de control del procedimiento analítico incluye los parámetros del procedimiento analítico que necesitan control y la prueba de idoneidad del sistema que forma parte de la descripción del procedimiento analítico. La descripción del procedimiento analítico debe incluir los pasos necesarios para realizar cada prueba analítica. Esto puede incluir (pero no se limita a) la muestra, los materiales de referencia y los reactivos, la preparación de la muestra y de control, el uso del aparato, la generación de la curva de calibración, el uso de las fórmulas con herramientas para el cálculo de los resultados notificables y otros pasos necesarios. El nivel de detalle debe permitir a un analista experto realizar el análisis e interpretar los resultados (como el nivel de detalle en una farmacopea regional para una sustancia similar). Esto se denomina comúnmente comprobación de la calidad de los datos. Se recomienda un seguimiento continuo de los resultados de los procedimientos analíticos seleccionados para buscar cualquier tendencia, de acuerdo con las expectativas. La revisión de los resultados de los procedimientos analíticos facilita la gestión del ciclo de vida del procedimiento y permite una intervención proactiva para evitar fallos.
El desafío en la revisión y mantenimiento del estado validado
La directriz ICH 14 establece de manera concisa la relación que existe entre el desarrollo de métodos, el proceso de validación y el mantenimiento del ciclo de vida con un enfoque en Calidad. Sin embargo, uno de los desafíos que enfrentamos en el día a día y llegado el momento es aquella que relaciona la obtención, disponibilidad y compilación de la información para realizar la revisión de estado validado. Sin duda la estructuración de herramientas informáticas que ayuden a mostrar y recuperar la información contenida un solo punto será de gran ayuda para dar cumplimiento a un proceso que requiere una comprensión absoluta del comportamiento y desempeño de un método analíticos durante todo su ciclo de vida.
Los alcances tecnológicos manifiestan avances de gestión que favorecen la mejora continua y el desempeño de las actividades de laboratorio.
La gestión y mantenimiento desde una perspectiva tecnológica se precisa de la siguiente manera:
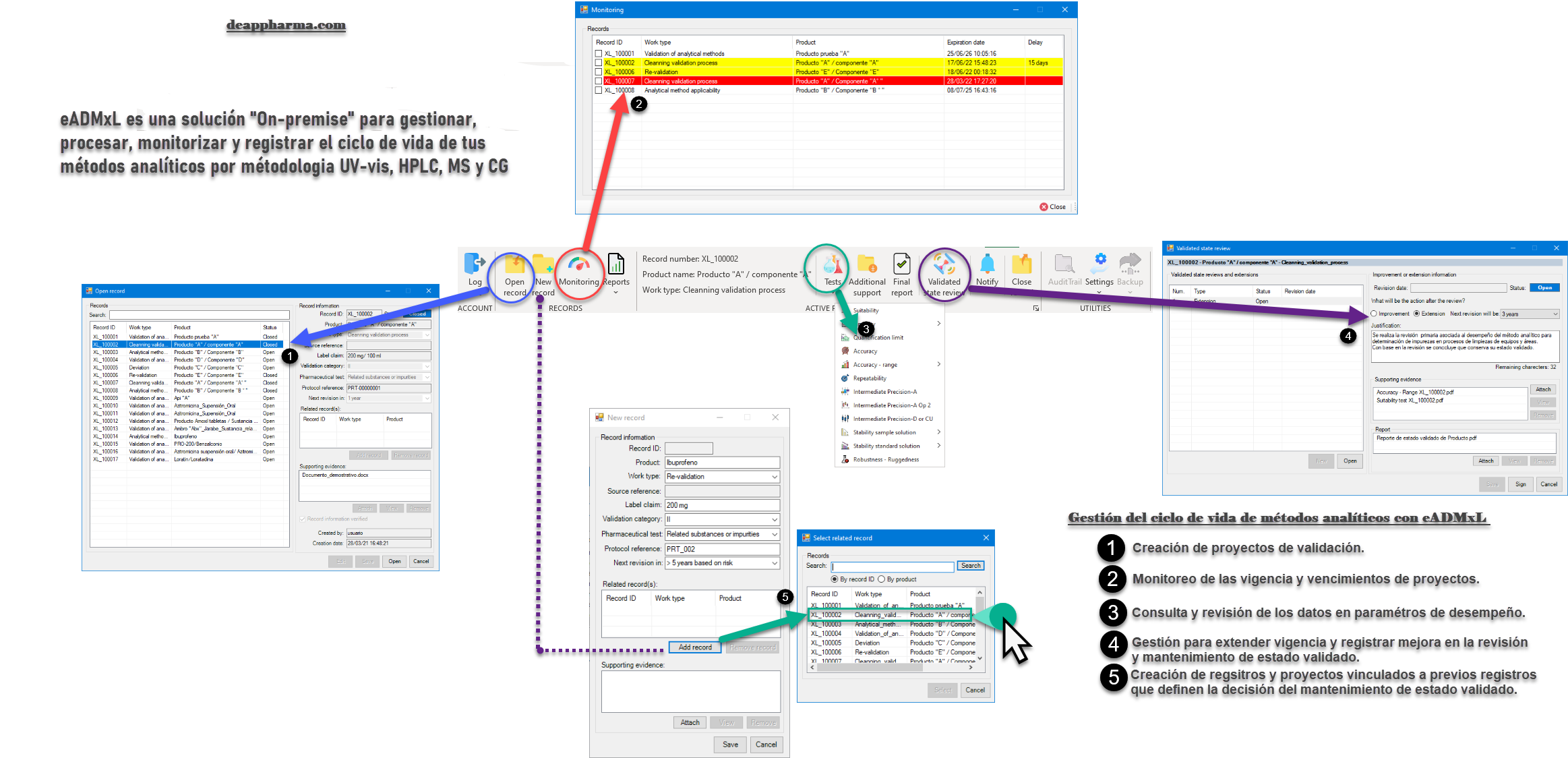
No olvides que, en caso de ser necesario, puedes contactarnos para mejorar el proceso de laboratorio y administrativo de tu organización a través software 100% confiable y seguro. Recuerda que en deappharma contamos con un software que te ayudara a controlar tu proceso de validación de métodos analíticos. Somos expertos en la materia, por lo que procesos complejos no representa un reto para nosotros, debido a que tenemos en mente los aspectos relevantes de cumplimiento.
Referencias
-
Analytical procedure development Q14 Draft version Endorsed on 24 March 2022
-
Validation of analytical procedures Q2(R2) Draft version Endorsed on 24 March 2022
¡Contáctanos! y conoce de que manera te vamos a ayudar.
¡Únete a nuestra comunidad en redes!