25, Feb 2022
¿Qué es un backup?
Entonces, definiremos como al backup, también llamado copia de respaldo o de seguridad, como la copia de la información sensible de un sistema informático desde su medio o medios de almacenamiento a un medio distinto de éste, sea cual fuere esta información, para su posterior almacenamiento en un entorno seguro con el supuesto fin de recuperar esta información con la mayor confiabilidad posible, si fuera necesario, en caso de que cualquier motivo o incidente genere la pérdida o modificación no deseada de los datos originales, para de esta manera obtener nuevamente, en un tiempo conveniente y razonable, la operatividad (consecuencia directa de la información) del sistema de cual se hizo la copia previa al incidente.
Cuando mencionamos que al realizar un backup copiamos la información sensible, nos referimos a los datos y metadatos que conforman el sistema informático. Es decir, que al realizar la copia de de la información con el objetivo de respaldarla no basta con copiar archivos, directorios o bases de datos, sino que los copiaremos con todas la características y cualidades, es decir, no sólo se trata de resguardar la información contenida en el archivo. Es importante mantener la estructura o imagen de estos al momento de realizar el backup, es decir, su ubicación (path), privilegios o permisos, si son de lectura, de sistema, etc.
Por otro lado, también mencionaremos el destino de esta copia, la cual debe hacerse a un medio distinto y en un entorno seguro. No nos serviría de nada generar una copia de la información en el mismo disco en el cual estamos trabajando o mantener el soporte destino junto al equipo. En ambos casos tanto la información original como el respaldo comparten los mismos riesgos, y esto es, precisamente lo que deseamos evitar.
Así mismo, el proceso se realiza presuponiendo cierta confiabilidad, pero no toda. En primer lugar, porque tampoco estamos exentos de que el soporte destino falle, éstos corren los mismos riesgos que los medios de origen, aunque en menor medida. En segundo lugar, porque puede existir un intervalo de tiempo significativo entre el día y la hora los cuales se ejecutó el proceso del backup y el día la hora en los cuales deseamos recuperar la información.
Por último, cuando mencionamos recuperar la información en un tiempo conveniente y razonable, nos referimos a la severidad con la que la información debe estar disponible nuevamente. Si bien la fiabilidad del backup es proteger nuestros datos, no nos sirve de mucho si los tiempos de recuperación son excesivos.
Si bien la tarea puede considerarse incomodo, y muchas veces sin sentido, nos evitará, sin ninguna duda, situaciones que hagan peligrar la operatividad de nuestros sistemas, y el ahorro de muchas situaciones irremediables.
Junto al termino backup aparece otro no menos importante: restore (recuperación o restauración), que es la operación inversa al backup. O sea, copiar al sitio original, desde el soporte destino, la información a la cual se le realizó backup anteriormente.
En muchas oportunidades se considera erróneamente backup a la copia de la información que se realiza en el mismo disco físico que contiene la información original, o tal vez se entiende que un nivel RAID es una especie de bakup, y no lo es. Si bien esta copia puede resultar útil en caso de modificaciones no deseadas o corrupción de la información del archivo o archivos originales, no nos permite recuperar la información en el caso de que la falla provenga del disco físico y nos provea estados del sistema anteriores al actual.
Por consiguiente, cuando hablamos de backup, nos referimos, no solo a copiar, sino pa hacerlo a un medio distinto del original. La idea conceptual del backup es extraer la información del sistema informático para ponerla a salvo, es decir, almacenarla en un entorno seguro. O, al menos, en un entorno que no comparta los mismos riesgos que el medio de origen.
No olvides que en caso de ser necesario, puedes contactarnos para mejorar el proceso de laboratorio y administrativo de tu organización a través software 100% confiable y seguro. Recuerda que en deappharma somos expertos en la materia, por lo que procesos complejos no representa un reto para nosotros, debido a que tenemos en mente los aspectos relevantes de cumplimiento.
Referencias
- Administración de Storage y Backups_Dante cantone, España, Editorial Alfa Omega
¡Contáctanos! y conoce de que manera te vamos a ayudar.
¡Únete a nuestra comunidad en redes!
- 0
- Por Team deappharma

11, Feb 2022
Audit Trail para asegurar y elevar el proceso de Integridad de datos
Este término regulatorio e informático se refiere a la auditoría, detección continua y exhaustiva de los datos. Por lo que es relevante contar con este proceso o función en cada uno de nuestros sistemas informatizados. El objetivo, es trazar la vida de un dato de manera perdurable.
La industria farmacéutica, dispositivos médicos y almacenes utilizan sistemas informatizados dentro de sus procesos. Por este hecho, es relevante el control y trazabilidad de datos. Para abordar el tema y conocer el alcance e importancia de la función de pistas de auditoria te invitamos a leer lo siguiente.
¿Qué es un “pista de auditoría”?
La «Pista de auditoría» o también conocida como «Audit Trail». Significa un registro electrónico seguro, generado por computadora con sellos de tiempo que permiten la reconstrucción del curso de los eventos relacionados con la creación, modificación y/o eliminación de un registro electrónico. Por ejemplo, la pista de auditoría para una ejecución de cálculos analíticos con la utilización de Excel como software de cálculo, debe incluir como mínimo lo siguiente: nombre de usuario, la fecha/hora de la ejecución, los cambios realizados sobre un valor y los detalles justificados de dicho cambio, según apliquen. ¡Pero no solo eso! Las pistas de auditoria son un aparte esencial de los sistemas computarizados. Es por esto, que los reportes de auditorita deben ser robustos y seguros.
Retomemos el ejemplo de uso de la hoja de Excel para obtención de resultados analíticos. En la actualidad la industria configura la obtención de pistas de auditoría sobre los mismos libros de Excel. Esta pista «LOG» regularmente la establecen dentro del mismo libro de manera anidada como una pestaña (hoja) de cálculo. ¡Esta práctica, es poco sostenible y segura! ya que la trazabilidad la dejas en manos de Excel y de la vulnerabilidad de código realizado con VBA.
¿Existe riesgo de perder la trazabilidad en mis libros de Excel por usar configuración VBA?
La respuesta es ¡Si! y tu impacto será alto porque no tendrás la manera de trazar la información.
Los riesgos los podemos clasificar en dos escenarios posibles:
- Daño en el archivo de Excel que no permita acceder a la información.
- Alteración y vulnerabilidad de la programación en VBA (Hasta videos en youtube te enseñan a desbloquear código relizadoo con VBA. ¡Ojo con esto!)
¿Quién debe revisar los registros de auditoría?
La revisión de la pista de auditoría es similar a la evaluación de las tachaduras en papel al revisar los datos. Personal responsable de la revisión de registros bajo cGMP debe revisar las pistas de auditoría que capturan los cambios en datos asociados con el registro a medida que revisan el resto del registro. Para ejemplo, todos los registros de producción y control, que incluyen registros de auditoría, deben revisarse y aprobado por la unidad de calidad. Las regulaciones brindan flexibilidad para tener algunas actividades revisadas por una persona que supervisa o verifica directamente la información.
FDA recomienda un enfoque de sistema de calidad para implementar la supervisión y revisión de cGMP registros.
¿Con qué frecuencia se deben revisar los registros de auditoría?
Si la frecuencia de revisión de los datos se especifica en las regulaciones de cGMP, adhiérase a esa frecuencia para la revisión de la pista de auditoría. Por ejemplo, requiere revisión después de cada paso significativo en fabricación, procesamiento, empaque o almacenamiento, y requiere la revisión de datos antes del lote liberar. En estos casos, aplicaría la misma frecuencia de revisión para la pista de auditoría.
Si la frecuencia de revisión de los datos no está especificada en las regulaciones de cGMP, debe determinar la frecuencia de revisión de la pista de auditoría utilizando el conocimiento de sus procesos y la evaluación de riesgos.
La evaluación de riesgos debe incluir la evaluación de la criticidad de los datos, los mecanismos de control e impacto en la calidad del producto. Los riesgos para los datos incluyen, entre otros, la posibilidad de que se eliminen modifique o excluyan sin autorización o sin detección.
Su enfoque para la revisión de la pista de auditoría y la frecuencia con la que la realiza deben garantizar que se cumplen los requisitos de cGMP, se implementan los controles apropiados y la confiabilidad de la revisión.
¿Puedo aumentar la trazabilidad de mis libros de Excel sin necesidad de usar VBA?
¡Si! y todo de manera virtual con el suso de nuestro complemento COM. Esto garantiza que tus datos sean trazados de manera perdurable.
En deappharma hemos desarrollado eDocuSeed una herramienta validada y especializada para el mantenimiento, gestión y trazabilidad de libros de Excel. ¡Sin duda te ayudaremos a mitigar tu riesgo! Puedes contactarnos para mejorar el proceso administrativo de tu organización a través software 100% confiable y seguro.
Referencias
- Data Integrity and Compliance With Drug CGMP Questions and Answers Guidance for Industry
- Guía para el enfoque de sistemas de calidad de la industria para las regulaciones CGMP farmacéuticas.
¡Contáctanos! y conoce de que manera te vamos a ayudar.
¡Únete a nuestra comunidad en redes!
24, Ene 2022
Detalles del Enfoque – Alcance de la Parte 11 de CFR
Detalles del Enfoque – Alcance de la Parte 11
Interpretación estrecha del alcance
Entendemos que existe cierta confusión sobre el alcance de la parte 11. Algunos han entendido que el alcance de la parte 11 es muy amplio. Creemos que algunas de esas amplias interpretaciones podrían conducir a controles y costos innecesarios y podrían desalentar la innovación y los avances tecnológicos sin brindar un beneficio adicional a la salud pública. Como resultado, queremos aclarar que la Agencia tiene la intención de interpretar el alcance de la parte 11 de manera restringida.
Según la interpretación restringida del alcance de la parte 11, con respecto a los registros que deben mantenerse según las reglas predicadas o presentarse a la FDA, cuando las personas optan por utilizar registros en formato electrónico en lugar de papel, se aplicaría la parte 11. Por otro lado, cuando las personas usan computadoras para generar impresiones en papel de registros electrónicos, y esos registros en papel cumplen con todos los requisitos de las reglas predicadas aplicables y las personas confían en los registros en papel para realizar sus actividades reguladas, la FDA generalmente no condicionara a las personas para «usar registros electrónicos en lugar de registros en papel». En estos casos, el uso de sistemas informáticos en la generación de registros en papel no activaría la parte 11.

Definición de registros de la Parte 11
Bajo esta interpretación estrecha, la FDA considera que la parte 11 es aplicable a los siguientes registros o firmas en formato electrónico (registros o firmas de la parte 11):
- Registros que deben mantenerse según los requisitos de la regla establecida y que se mantienen en formato electrónico en lugar del formato en papel. Por otro lado, los registros (y cualquier firma asociada) que no se requiera conservar según las reglas establecida, pero que, sin embargo, se mantienen en formato electrónico, no son registros de la parte 11.
Le recomendamos que determine, en función de las reglas establecidas, si los registros específicos son registros de la parte 11. Le recomendamos que documente tales decisiones.
- Registros que deben mantenerse según las reglas establecidas, que se mantienen en formato electrónico además del formato en papel, y en los que se confía para realizar actividades reguladas.
En algunos casos, las prácticas comerciales reales pueden dictar si está utilizando registros electrónicos en lugar de registros en papel. Por ejemplo, si se requiere que se mantenga un registro bajo una regla predicada y usted usa una computadora para generar una copia impresa en papel de los registros electrónicos, pero aun así depende del registro electrónico para realizar actividades reguladas, la Agencia puede considerar que usted está utilizando el registro electrónico en lugar del registro en papel. Es decir, la Agencia puede tener en cuenta sus prácticas comerciales para determinar si se aplica la parte 11.
En consecuencia, recomendamos que, para cada registro que deba mantenerse según las reglas establecidas, determine de antemano si planea confiar en el registro electrónico o en el registro en papel para realizar actividades reguladas. Recomendamos que documente esta decisión (por ejemplo, en un Procedimiento operativo estándar (SOP) o documento de especificación).
Registros enviados a la FDA, conforme a reglas establecidas (incluso si dichos registros no están específicamente identificados en las reglamentaciones de la Agencia) en formato electrónico. Sin embargo, un registro que no se presenta en sí mismo, pero que se utiliza para generar una presentación, no es un registro de la Parte 11 a menos que se requiera que se mantenga bajo una regla establecida y se mantenga en formato electrónico.
Firmas electrónicas que pretenden ser el equivalente de firmas manuscritas, iniciales y otras firmas generales requeridas por reglas establecidas. Las firmas de la Parte 11 incluyen firmas electrónicas que se utilizan, por ejemplo, para documentar el hecho de que ciertos eventos o acciones ocurrieron de acuerdo con la regla establecida (por ejemplo, aprobado, revisado y verificado).
Enfoque de los requisitos específicos de la Parte 11
Validación
La Agencia tiene la intención de ejercer la discreción de cumplimiento con respecto a los requisitos específicos de la parte 11 para la validación de sistemas computarizados y los requisitos correspondientes. Aunque las personas aún deben cumplir con todos los requisitos de las reglas establecidas aplicables para la validación, esta guía no debe interpretarse como una imposición de requisitos adicionales para la validación.
Sugerimos que su decisión de validar los sistemas computarizados y el alcance de la validación tengan en cuenta el impacto que los sistemas tienen en su capacidad para cumplir con los requisitos de las reglas establecidas. También debe considerar el impacto que esos sistemas podrían tener en la precisión, confiabilidad, integridad, disponibilidad y autenticidad de los registros y firmas requeridos. Incluso si no existe un requisito de regla de establecido para validar un sistema, en algunos casos puede ser importante validar el sistema.
Recomendamos que base su enfoque en una evaluación de riesgos justificada y documentada y una determinación del potencial del sistema para afectar la calidad y seguridad del producto y la integridad del registro. Por ejemplo, la validación no sería importante para un procesador de texto que se usa solo para generar SOP.
Pista de auditoría
La Agencia tiene la intención de ejercer la discreción de cumplimiento con respecto a los requisitos específicos de la parte 11 relacionados con las pistas de auditoría generadas por computadora y con sello de tiempo y cualquier requisito correspondiente. Las personas aún deben cumplir con todos los requisitos de reglas establecidas aplicables relacionados con la documentación de, por ejemplo, fecha, hora o secuencia de eventos, así como cualquier requisito para garantizar que los cambios en los registros no oscurezcan entradas previas.
Incluso si no existen requisitos de reglas establecidas para documentar, por ejemplo, la fecha, la hora o la secuencia de eventos en una instancia particular, puede ser importante contar con pistas de auditoría u otras medidas de seguridad físicas, lógicas o de procedimiento para garantizar la confiabilidad y confiabilidad de los registros. Le recomendamos que base su decisión de aplicar pistas de auditoría u otras medidas apropiadas, en la necesidad de cumplir con los requisitos de la regla establecida, una evaluación de riesgos justificada y documentada, y una determinación del efecto potencial sobre la calidad y seguridad del producto y la integridad del registro. Le sugerimos que aplique los controles apropiados basados en dicha evaluación. Los registros de auditoría pueden ser particularmente apropiados cuando se espera que los usuarios creen, modifiquen o eliminen registros regulados durante el funcionamiento normal.
Sistemas heredados
La Agencia tiene la intención de ejercer la discreción de ejecución con respecto a todos los requisitos de la parte 11 para los sistemas que de otro modo estaban operativos antes del 20 de agosto de 1997, la fecha de vigencia de la parte 11, bajo las circunstancias especificadas a continuación.
Esto significa que la Agencia no tiene la intención de tomar medidas de cumplimiento para imponer el cumplimiento de los requisitos de la parte 11 si se cumplen todos los siguientes criterios para un sistema específico:
- El sistema estaba operativo antes de la fecha de vigencia.
- El sistema cumplió con todos los requisitos de reglas establecidas aplicables antes de la fecha de vigencia.
- Actualmente, el sistema cumple con todos los requisitos de reglas establecidas aplicables.
- Tiene evidencia documentada y justificación de que el sistema es apto para el uso previsto (lo que incluye tener un nivel aceptable de seguridad e integridad de registros, si corresponde).
Si un sistema ha cambiado desde el 20 de agosto de 1997, y si los cambios impiden que el sistema cumpla con los requisitos de la regla establecidad, los controles de la Parte 11 deben aplicarse a los registros y firmas de la Parte 11 de conformidad con la política de cumplimiento expresada en esta guía.
Copias de Registros
La Agencia tiene la intención de ejercer la discreción de ejecución con respecto a los requisitos específicos de la parte 11 para generar copias de registros y cualquier requisito correspondiente. Debe proporcionar a un investigador un acceso razonable y útil a los registros durante una inspección. Todos los registros en su poder están sujetos a inspección de acuerdo con las reglas establecidas.
Recomendamos que proporcione copias de registros electrónicos por:
- Producir copias de registros mantenidos en formatos portátiles comunes cuando los registros se mantienen en estos formatos
- Usar métodos de conversión o exportación automatizados establecidos, cuando estén disponibles, para hacer copias en un formato más común (los ejemplos de dichos formatos incluyen, entre otros, PDF, XML o SGML)
En cada caso, recomendamos que el proceso de copiado produzca copias que conserven el contenido y el significado del registro. Si tiene la capacidad de buscar, ordenar o buscar tendencias en los registros de la parte 11, las copias entregadas a la Agencia deben proporcionar la misma capacidad si es razonable y técnicamente factible. Debe permitir la inspección, revisión y copia de registros en un formato legible en su sitio utilizando su hardware y siguiendo sus procedimientos y técnicas establecidos para acceder a los registros.
Retención de registros
La Agencia tiene la intención de ejercer la discreción de ejecución con respecto a los requisitos de la parte 11 para la protección de registros para permitir su recuperación precisa y rápida durante todo el período de retención de registros y cualquier requisito correspondiente. Las personas aún deben cumplir con todos los requisitos de reglas establecidas aplicables para la retención y disponibilidad de registros.
Sugerimos que su decisión sobre cómo mantener los registros se base en los requisitos de las reglas predicadas y que base su decisión en una evaluación de riesgos justificada y documentada y en una determinación del valor de los registros a lo largo del tiempo.
FDA no tiene la intención de objetar si usted decide archivar los registros requeridos en formato electrónico en medios no electrónicos como microfilm, microficha y papel, o en un formato de archivo electrónico estándar (ejemplos de tales formatos incluyen, entre otros, PDF, XML o SGML). Las personas aún deben cumplir con todos los requisitos de las reglas establecidas, y los propios registros y cualquier copia de los registros requeridos deben conservar su contenido y significado. Siempre que los requisitos de la regla establecida se cumplan por completo y el contenido y el significado de los registros se conserven y archiven, puede eliminar la versión electrónica de los registros. Además, los componentes de firmas y registros en papel y electrónicos pueden coexistir (es decir, una situación híbrida) siempre que se cumplan los requisitos de la regla establecida y se conserve el contenido y el significado de esos registros.
No olvides que en caso de ser necesario, puedes contactarnos para mejorar el proceso de laboratorio y administrativo de tu organización a través software 100% confiable y seguro. Recuerda que en deappharma somos expertos en la materia, por lo que procesos complejos no representa un reto para nosotros, debido a que tenemos en mente los aspectos relevantes de cumplimiento.
REFERENCES
- Glossary of Computerized System and Software Development Terminology (Division of Field Investigations, Office of Regional Operations, Office of Regulatory Affairs, FDA 1995)
- General Principles of Software Validation; Final Guidance for Industry and FDA Staff (FDA, Center for Devices and Radiological Health, Center for Biologics Evaluation and Research, 2002)
- Guidance for Industry, FDA Reviewers, and Compliance on Off-The-Shelf Software Use in Medical Devices (FDA, Center for Devices and Radiological Health, 1999)
- Pharmaceutical CGMPs for the 21st Century: A Risk-Based Approach; A Science and Risk-Based Approach to Product Quality Regulation Incorporating an Integrated Quality Systems ApproachExternal Link Disclaimer (FDA 2002)
¡Contáctanos! y conoce de que manera te vamos a ayudar.
¡Únete a nuestra comunidad en redes!
13, May 2021
Automatización de la gestión documental.
En los últimos años, un gran número de organizaciones ha implementado procesos documentales que evidencian y soportan las funciones realizadas como resultado de sus actividades diarias. Dichos procesos son de carácter secuencial y transversal a las mismas, y están destinados a la planeación, producción, gestión y trámite, organización, transferencia, disposición, preservación y valoración de los documentos, ya sean físicos o electrónicos. Basado en lo anterior, la gestión se realiza, generalmente, a través de la producción de documentos físicos, haciendo uso de planillas de entrega, formatos de préstamos documentales, acuses de recibido, entre otros; y documentos electrónicos como correos electrónicos e información no estructurada que, dependiendo de cada organización, puede ser ejecutada por humanos o por sistemas especializados para tal fin.
En este sentido, una organización produce un extenso número de documentos que, sin una adecuada gestión documental, pueden generar problemas como desperdicio de papel, pérdida de información, gran cantidad de tiempo en consulta o recuperación de información, entre muchos otros inconvenientes administrativos y costes económicos para de la entidad. Para contrarrestar y mitigar este tipo de situaciones, se hace necesaria la implementación de la automatización para la transformación de la gestión documental, entendida como la capacidad de hacer uso de sistemas para llevar a cabo ciertas acciones que normalmente son ejecutadas por personas, soportado con diferentes metodologías y tecnologías.
La automatización de documentos (también conocido como ensamblaje de documentos) es el diseño de sistemas y flujos de trabajo que ayudan en la creación de documentos electrónicos. Estos incluyen sistemas basados en la lógica que utilizan segmentos de texto o información ya existentes para generar un nuevo documento. La automatización de documentos puede ser utilizada para automatizar todo texto condicional, texto variable e información contenidos en un conjunto de documentos.
Los sistemas de automatización permiten a las empresas reducir al mínimo la entrada de datos, el tiempo dedicado a la lectura y corrección, así como los errores humanos. Algunos beneficios adicionales pueden ser: ahorro de tiempo y financiero debido al menor manejo de papel, carga de documentos, almacenamiento, distribución, envío, trabajo y gasto.
En tal razón, la automatización de procesos documentales es un factor clave en la transición de la gestión documental manual a la realizada de forma automática o semiautomática por medio de tecnologías de información.
De esta manera, se puede determinar que la automatización de procesos documentales conlleva una serie de oportunidades para las organizaciones como lo son: reducción de costos, programación automática de tareas, control y seguimiento de estas en tiempo real permitiendo la generación de reportes; disminución de tiempo que deriva en aumento de la productividad y evitando reprocesos por errores operativos. Todo esto, en conformidad con los marcos regulatorios existentes soportados en las políticas “cero papel”, promoviendo la normalización de los archivos en las organizaciones y generando poco a poco una cultura digital.
Beneficios primarios.
Control de versiones.
El auge de los sistemas de gestión documental ha supuesto la mejora y actualización de numerosas funcionalidades. El control de versiones ha estado durante muchos años ligado a la informática favoreciendo la labor de los desarrolladores, pero ¿qué labor puede realizar para mejorar la gestión documental?
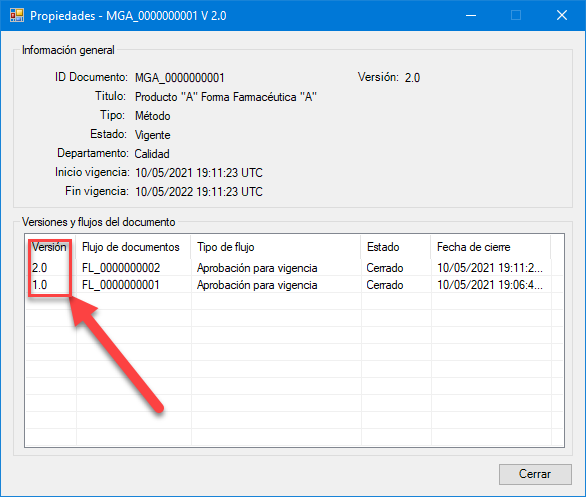
¿Qué es el control de versiones?
Se llama control de versiones a la gestión de cambios efectuados en un documento y otros archivos que contengan información. Los cambios se registran de forma automática y pueden ser identificados mediante números o combinaciones alfanuméricas. En los sistemas de gestión documental con funciones avanzadas de control de versiones, cada cambio no señalará únicamente las modificaciones efectuadas, sino quién y cuándo se realizaron.
Historial de versiones.
El historial de versiones recoge en una misma localización todas las versiones que se han creado de un mismo documento. Es una característica muy diferente dependiendo del tipo de gestor documental con el que se trabaje. Por ejemplo, en los sistemas más avanzados el control de versiones establece la posibilidad de generar un historial ilimitado de versiones al que se podrá volver siempre que se crea necesario.
Seguimiento de cambios.
El seguimiento de cambios es el concepto entorno al que gira la idea del control de versiones. Dependiendo del sistema que se utilice y de lo avanzado de sus características, esta funcionalidad determinará aspectos diferentes.
En los sistemas de gestión documental en los que la colaboración en documentos es un aspecto recurrente, el seguimiento de cambios es una característica fundamental. Por ejemplo, en un documento en el que trabajan diferentes empleados, cada uno aporta parte de sus conocimientos al mismo. Con el seguimiento de cambios no se corre el riesgo de pérdida o modificación de la información, ya que siempre se permite volver a la versión anterior de cualquier archivo.
No olvides que en caso de ser necesario, puedes contactarnos para mejorar el proceso de laboratorio y administrativo de tu organización a través software 100% confiable y seguro. Recuerda que en deappharma somos expertos en la materia, por lo que procesos complejos no representa un reto para nosotros, debido a que tenemos en mente los aspectos relevantes de cumplimiento.
¡Contáctanos! y conoce de que manera te vamos a ayudar.
¡Únete a nuestra comunidad en redes!