23, Ene 2024
Requerimientos generales para la validación de sistemas computarizados
Cuando hablamos de sistemas computarizados debemos tener en cuenta ciertos principios de validación que pueden repercutir la calidad de los resultados, el control de documentos y el almacenamiento de datos. El objetivo de la validación es garantizar la confianza en los datos de laboratorio capturados, procesados, notificados o almacenados por los sistemas informatizados. Un sistema validado garantiza la exactitud de los resultados y reduce los riesgos para la integridad de los datos. A continuación, abordaremos los requerimientos generales establecidos por la OMCL (Official Medicines Control Laboratories) de la unión europea (EDQM) para la validación de sistemas computarizados.
Inventario
Se debe mantener un inventario o una lista equivalente debe estar disponible. La información mínima que debe incluir el inventario de sistemas computarizados, es:
-Identificación y versión
-Propósito
-Estatus de la validación
-Tipo de almacenamiento
-Persona de contacto
Validación
Antes de su uso rutinario, el sistema informatizado deberá ser validado.
El objetivo de la validación es confirmar que las especificaciones del sistema informatizado se ajustan a las necesidades del usuario y los usos previstos mediante el examen y la aportación de pruebas objetivas y que los requisitos particulares pueden cumplirse de forma coherente.
El alcance de la validación dependerá de la complejidad y del uso previsto del sistema informatizado que se valide.
El esfuerzo de validación puede escalarse y adaptarse al tipo de sistema justificado por una evaluación de riesgos documentada.
Registro de problemas
Debe mantenerse un registro de los problemas identificados por los usuarios y de las medidas adoptadas.
Control de cambios
En caso de cambios en el sistema informático, incluidas las actualizaciones de versión, lo ideal sería en un entorno de prueba, tras lo cual deberá restablecerse el estado de validación.
Si es necesaria una revalidación, debe realizarse no sólo para validar el cambio individual, sino
también para determinar el alcance y el impacto de ese cambio en todo el sistema informatizado.
El alcance de la revalidación dependerá de la evaluación del cambio o cambios, que deberá estar
documentada. Un posible enfoque podría ser el uso de cuadernos de bitácora como se hace para los equipos, y/o la utilización de un procedimiento documentado de control de cambios.
Revisión periódica
El OMCL debe adoptar una política de comprobación periódica del sistema informatizado para evitar cualquier error y garantizar el mantenimiento del estado de validación. La frecuencia de las revisiones deberá definirse en función de los riesgos. Los sistemas informatizados deberán estar cubiertos por la estrategia de auditoría interna.
Seguridad y condiciones ambientales
Los sistemas informatizados deben estar protegidos contra cualquier intrusión que pueda modificar los datos y afectar a los resultados finales.
Las salas de servidores deben tener acceso restringido y contar con las condiciones necesarias para garantizar el correcto funcionamiento de los equipos (control de temperatura, medidas contra incendios, Sistema de Alimentación Ininterrumpida, etc.).
El acceso a los sistemas debe estar restringido al personal autorizado, mediante cuentas personalizadas y contraseña o método de identificación equivalente. Debe evitarse el uso de cuentas compartidas y genéricas para garantizar que las acciones documentadas en los sistemas informáticos puedan atribuirse a una única persona. Cuando no se disponga de cuentas personales o éstas no sean viables, deberán utilizarse combinaciones de registros en papel y electrónicos para rastrear las acciones del personal responsable.
Sólo la(s) persona(s) responsable(s) o el personal informático designado deben tener derechos administrativos para implementar cualquier actualización y/o instalación de sistemas informáticos, cambiar ajustes críticos del sistema (por ejemplo, registro de auditoría, hora/fecha) y gestionar los permisos de otros usuarios. Todas las tareas rutinarias, como las de análisis, deben basarse en una cuenta de usuario y una contraseña que no tengan derechos administrativos.
Los derechos administrativos deben documentarse y concederse únicamente al personal con funciones de mantenimiento del sistema (por ejemplo, informáticos) que sean totalmente independientes del personal responsable del contenido de los registros (por ejemplo, analistas de laboratorio, dirección del laboratorio).
Cuando no sea posible asignar de seguridad independientes, deberán utilizarse otras estrategias de control para reducir los riesgos para la integridad de los datos.
Los ordenadores deben bloquearse después de su uso y no debe permitirse a los usuarios cambiar los ajustes de fecha y hora.
El hardware utilizado debe cumplir los requisitos técnicos para que el trabajo se pueda realizar. Dichos requisitos incluyen, por ejemplo, los requisitos mínimos del sistema indicados por el fabricante del sistema. Estos requisitos deben estar predefinidos en función del uso previsto.
Los componentes de hardware deben ser instalados por personal cualificado (por ejemplo, personal de la Unidad de Tecnologías de la Información (TI), un técnico del fabricante del equipo, etc.).
Registro de auditoría
El sistema informático debe mantener un registro de todas las acciones críticas que se produzcan, por ejemplo, quién ha accedido a él y cuándo, cualquier supresión o modificación de datos, etc. Si un sistema informatizado no registra automáticamente una pista de auditoría, el OMCL deberá mantener un registro alternativo.
No se permitirá a los usuarios modificar o desactivar las pistas de auditoría o los medios alternativos de proporcionar trazabilidad de las acciones de los usuarios.
En todos los sistemas informatizados nuevos deberá tenerse en cuenta la necesidad de implantar una función de pista de auditoría adecuada.
Cuando un sistema informatizado existente carezca de pistas de auditoría generadas por ordenador, el personal utilizará medios alternativos como el uso controlado por procedimientos de libros de registro, control de cambios, control de versiones de registros u otras combinaciones para cumplir el el requisito de trazabilidad para documentar el qué, quién, cuándo y por qué de una acción.
Firmas electrónicas
Si se utilizan firmas electrónicas, una declaración sobre la equivalencia de la firma electrónica a la firma manuscrita o declaración legal similar debe existir dentro del sistema de gestión de la calidad.
Archivo de versiones sustituidas de sistemas informáticos
Las versiones sustituidas de los programas informáticos deben archivarse de forma recuperable si es necesario para acceder a datos históricos, de acuerdo con la directriz de la OMCL «Gestión de documentos y registros».
Para el caso de los programas informáticos comerciales, la obligación de archivar las versiones anteriores puede estar sujeta al contrato con el proveedor.
Formación
Se garantizará la correcta utilización y validación del sistema informático. Esto puede hacerse mediante una formación adecuada y documentada o mediante información detallada en los procedimientos pertinentes o información contextual en el programa informático.
La formación se impartirá antes del primer uso y después de cada cambio importante en el software (por ejemplo, actualización de la versión). Las personas responsables de la validación recibirán formación sobre el proceso de validación.
¡Recuerda! que en deappharma tienes un aliado para realizar la validación de sistemas computarizados. Con nuestro método obtendrás el cumplimiento requerido.
Con nuestro servicio obtendrás un beneficio totalmente gratis por un año. Este beneficio consiste en implementar eDocuSeed un software que centraliza todos tus libros de Excel en un solo punto. Para brindarles los atributos de integridad de datos, trazabilidad y mantenimiento necesarios para dar cumplimiento a las regulaciones más exigentes. Nuestra solución ya impacta de manera positiva a empresas del sector farmacéutico, dispositivos médicos y distribuidoras de medicamentos. Solicita tu demostración totalmente gratuita. ¡Estamos listos para ayudarte!
Contáctanos y solicita una demostración y asesoría gratuita ¡Quiero un asesoría y demostración!
¡Gracias por leernos!
- 0
- Por Team deappharma

3, Jul 2023
Hojas de cálculo; categorización conforme a GAMP 5
Las hojas de cálculo pueden ser tan complejas o simples según la necesidad de la actividad a realizar. Sin embargo, al momento de evaluar una hoja de cálculo el proceso es mucho más sistemático de lo que se cree. Te invitamos a leer nuestro siguiente articulo y conoce algunos detalles relevantes que te ayudaran a categorizar tu hoja de cálculo.
Enfoque basado en el riesgo
Una aplicación de hoja de cálculo puede variar significativamente en cuanto a riesgo y complejidad. No obstante, en todos los casos es necesario cumplir los siguientes requisitos:
- Evaluación del riesgo y medidas de control del riesgo apropiadas para gestionar el riesgo identificado.
- Especificación y verificación apropiadas para demostrar que la aplicación funciona según lo previsto.
La estrategia de especificación y verificación de la aplicación que se está construyendo debe basarse en:
- El impacto del sistema en la seguridad del paciente, la calidad del producto y la integridad de datos mediante la evaluación del riesgo.
- Complejidad y novedad del sistema (arquitectura y categorización de los componentes del sistema).
- La seguridad apropiada para mitigar el riesgo de cambios no autorizados en los datos o la aplicación.
Es importante mencionar que el proceso de validación de sistemas computarizados en los cuales están incluidas las hojas de cálculo deben estar regidas por políticas y procedimientos de la empresa para definir y lograr mantener la conformidad y la idoneidad para el uso previsto de la aplicación por el usuario final.
Uso de categorías GAMP 5
La categorización de hojas de cálculo se debe establecer con base en la guía GAMP 5. La guía nos proporciona lineamiento y algunos ejemplos generales que nos ayudan a comprender el rumbo a tomar para realizar el proceso de validación de este tipo de archivos.
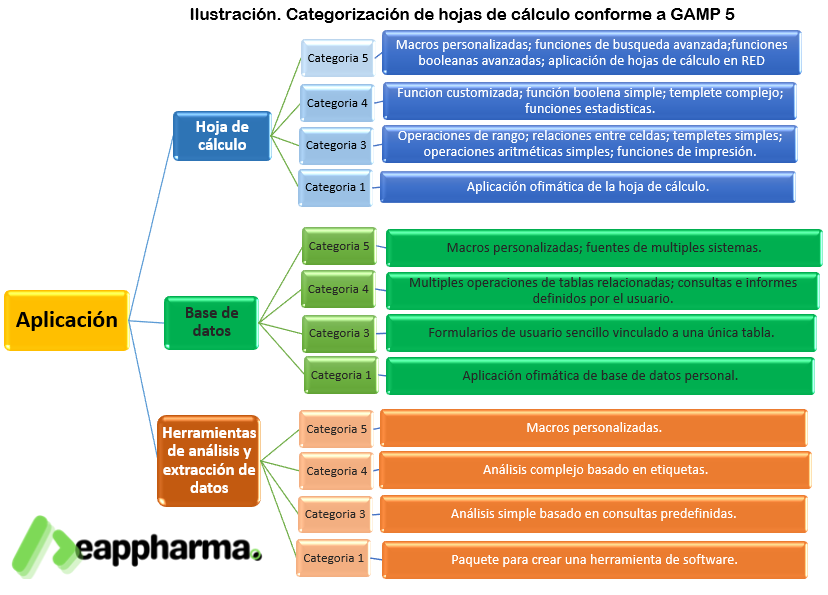
GAMP 5 establece y define 4 categorías de clasificación siendo las siguientes; 1, 3, 4 y 5. A continuación se describen criterios y detalles con base en esta Guía.
La herramienta sobre la que se construye la aplicación, como el paquete de hoja de cálculo, debe considerarse como categoría 1.
Las categorías para hojas de cálculo y otras aplicaciones de usuario final deben considerarse como un continuo que abarca las categorías 3, 4 y 5. La asignación de una categoría depende de la complejidad y personalización de la hoja de cálculo o aplicación. No obstante, hay que tener en cuenta que una hoja de cálculo que se limita a utilizar la capacidad de esta para edición, tabulación y no realiza ningún cálculo debe considerarse un documento. El uso previsto de los datos ensayados debe tenerse en cuenta a la hora de determinar los requisitos de verificación y el control que debe establecerse. Una simple hoja de cálculo (categoría 3) puede presentar un riesgo BxP (o GxP) elevado en función del uso que se haga de los datos.
Una hoja de cálculo que simplemente utiliza funciones nativas para realizar cálculos en lugar de una calculadora de mano suele ser de categoría 3. Por ejemplo, un analista de laboratorio puede crear una hoja de cálculo única para realizar un cálculo relacionado con una investigación derivado de un fuera de especificación. Cuando se utiliza la función aritmética de las hojas de cálculo, el cálculo debe explicarse detalladamente. Los datos deben explicarse de la misma manera que en un documento de texto. Esto debe incluir la verificación de que el cálculo se ha realizado correctamente y que los datos analizados son los correctos. Esta verificación podría documentarse fácilmente haciendo que otro analista o un supervisor examinara la hoja de cálculo y la aprobara. No se requiere ninguna otra verificación, ya que no es necesario cuestionar la exactitud del cálculo.
Cuando se desarrollen hojas de cálculo como templetes, podrían ser de categoría 3 a 5, dependiendo de la complejidad. Para el propósito de la aplicación de usuario, la definición de la categoría 4 se altera ya que esta se centra en la complejidad frente a la configurabilidad.
Por ejemplo:
- Un analista utiliza un templete en el laboratorio para realizar un cálculo rutinario de medias y desviación estándar de resultados experimentales. Se trata de una operación aritmética directa sin configuración, por lo que el templete es de categoría 3.
- Un templete de hoja de cálculo requiere que el usuario introduzca la dureza de la tableta, por lo que la aplicación se ramifica automáticamente a diferentes celdas (customs) para utilizar el cálculo específico de la dureza basado en esta entrada. Una operación tan simple haría que la hoja fuera de categoría 4, ya que tiene alguna operación booleana (es decir, celdas vinculantes o condicionales lógicas a otras celdas para procesar datos de entrada) simple basada en la entrada del usuario.
- Una aplicación de hoja de cálculo que emplee macros personalizadas o una lógica anidada sofisticada para obtener resultados o una función de búsqueda debería tratarse como categoría 5.
En la siguiente ilustración podrás observar el alcance generar por aplicación y la relación que estas tienen respecto a sus diferentes categorías.
¿Existen herramientas en el mercado que mejoren los procesos de control y mantenimiento de libros de Excel?
Como bien sabemos el desafío más grande al que se ha puesto a prueba el sector farmacéutico es el relacionado con la integridad de datos. Derivado de la gran cantidad de datos emitidos día con día en el que se ven impactados documentos y libros de Excel.
Recordemos que la validación de hojas de cálculo es el primer paso. No obstante, muchos libros de Excel que contienen hojas de cálculo utilizados en el sector farmacéutico no cuentan con evidencia de validación lo cual es un riesgo que podrá derivar en un hallazgo critico ante una inspección regulatoria. El segundo paso y el más complejo es adherir las configuraciones necesarias que eleven la integridad de datos y favorezcan la gestión y el mantenimiento de estas con base en GAMP 5 y CFR21.
En la actualidad el trabajo realizado y el menos sugerible es la configuración de control de seguridad y trazabilidad sobre usos de hojas de cálculo con el uso de VBA lo cual es meramente simbólico que a mediano y largo plazo será poco sostenible en términos de cumplimiento e integridad de datos.
En deappharma tenemos la solución para mitigar tu riesgo por el uso de hojas de cálculo no validadas y con deficiencias en los atributos de control de integridad de datos. Hemos logrado automatizar el proceso y, con esto ahorres tiempo, dinero y esfuerzo para dar el cumplimiento regulatorio requerido. Nuestros desarrollos están estructurados bajo un sistema de gestión de calidad trazable y auditable y, tales cuales cuentan con el soporte de validación-calificación que garantizan la precisión del alcance del diseño establecido.
Estamos convencidos de que eDocuSeed te ayudara a mitigar el uso de hojas de cálculo y dar ese paso de cumplimiento que necesitas. Conoce sus beneficios y alcance general aquí: ¡Da clic aquí!
“No vivas con el riesgo ¡Mitígalo!” para eso nosotros de ayudamos.
Visita nuestro sitio www.deappharma.com y solicita una demostración y asesoría gratuita.
¡Contáctanos y únete a nuestra comunidad en redes!
21, Jun 2023
Hojas de cálculo; tipificación conforme a GAMP 5
Tipificación de aplicaciones para hojas de cálculo
Las hojas de cálculo son herramientas de uso cotidiano en el sector regulado. Por este hecho, estas herramientas deben cumplir con los requerimientos descritos por la guía GAMP 5 (Good Automated Manufacturing Practices) en su versión vigente. Como bien sabemos la versatilidad de Excel proporciona una alta gama de funciones que favorecen la personalización de este tipo de archivos y por este hecho el riesgo asociado a la salud del paciente, calidad del producto e integridad de datos pueden ser potencialmente afectado.
En la actualidad los eventos detectados sobre la integridad de datos son cada vez más recurrentes ya que las auditorias se realizan con base en riesgos. Los hallazgos de integridad de datos son sobre el sistema documental en el cual de manera implícita están involucradas las hojas de cálculo las cuales para su gestión terminan siendo un desafío.
Cuando trabajamos con hojas de cálculo debemos comprender que el cumplimiento de estas y el alcance de la validación depende del grado de complejidad e impacto que esta tenga dentro de los procesos BxP (ó GxP). Lo anterior no quiere decir que hojas de cálculo de complejidad e impacto bajo estén exentas del control y su mantenimiento.
En nuestro siguiente artículo podrás consultar información que te ayudara a comprender maneras habituales y tipificaciones de usos de hojas de cálculo con base en la GAMP 5.
Hojas de cálculo desechables (uso como calculadora)
Las hojas de cálculo pueden utilizarse del mismo modo que una calculadora de mano. Por ejemplo, se introducen 10 valores de salida de una prueba de laboratorio para calcular la media y la desviación estándar. En este caso, no se conserva la copia electrónica.
Esto debe documentarse de la misma forma que se documentaría el uso de una calculadora sin impresora. Los resultados pueden imprimirse, etiquetarse y firmarse. Como alternativa, pueden guardarse en un formato estático y firmarse mediante una herramienta externa de firma electrónica. Debe quedar claro en la página de manipulación aritmética lo que se ha realizado exactamente. Esto puede facilitarse en la mayoría de las herramientas de hojas de cálculo imprimiendo una copia de las hojas de cálculo que muestre las fórmulas de las celdas. Esto no significa que haya que comprobar la exactitud de los algoritmos utilizados por las funciones nativas de la hoja de cálculo cada vez que se ejecute la hoja, sino demostrar que son los cálculos correctos durante la fase de verificación. Por ejemplo, a+b/c es una expresión muy diferente de (a+(b/c)), y es fácil cometer errores de este tipo. La verificación del cálculo puede realizarse imprimiendo las fórmulas de las celdas, o mediante un revisor externo. Esta verificación de los cálculos es apropiada para cualquier hoja de cálculo BxP (ó GxP).
Hojas de cálculo conservada como documento
En muchos casos, la forma en que se utilizan las hojas de cálculo es más parecida a la de un documento de tratamiento de textos que a la de una aplicación tridimensional. La principal diferencia es que las hojas de cálculo pueden utilizarse tanto para registrar datos BxP (ó GxP) como para manipularlos. La flexibilidad de manipulación que hace que las hojas de cálculo sean útiles aconseja gestionarlas como documentos y no como aplicaciones. Es probable que resulte extremadamente difícil que todas las copias guardadas subsecuentes son iguales al original. Por tanto, los cálculos deben verificarse y explicarse detalladamente, como se haría en un documento textual. Esto debería incluir la prueba de que se han utilizado las fórmulas correctas.
El nivel de riesgo relacionado con la integridad de datos debe tenerse en cuenta a la hora de elegir una estrategia de control. Existe una variedad de opciones para lograr un control adecuado, como:
- Utilizar la opción de seguridad interna de las herramientas de hoja de cálculo, como la protección de celdas u hojas mediante contraseña.
- Almacenamiento de la hoja de cálculo en un directorio seguro.
- Modificar las hojas de cálculo dentro de un EDMS (Electronic Document Management Software) como lo es eDocuSeed.
Si la hoja de cálculo no puede controlarse adecuadamente a través de estos u otros medios, puede ser aconsejable considerar una versión estática, por ejemplo, un PDF seguro o incluso una copia impresa, como registro primario. Sin embargo, por la gran cantidad de hojas de cálculo que la industria farmacéutica genera y usa este método de control se vuelve insostenible y riesgoso en términos de atributos de integridad de datos. No obstante, los EDMS robustos y de alto cumplimiento como lo es eDocuSeed brindan los controles de gestión y mantenimiento necesarios para archivos de hojas de cálculo para evitar con esto evitar el control impreso.
Las hojas de cálculo que sean efectivamente documentos deben gestionarse de conformidad con la normativa aplicable en términos de buenas prácticas de documentación. Por ejemplo, un uso común de las hojas de cálculo es manipular y mantener datos de laboratorio BxP (ó GxP), donde el cumplimiento de la normativa sobre registros y firmas electrónicas es una preocupación particular.
Hojas de cálculo como base de datos
Otro uso popular de las hojas de cálculo es como base de datos sencilla, por ejemplo, para gestionar o almacenar datos de forma electrónica. En un entorno BxP (ó GxP), esto supone un riesgo porque los datos pueden actualizarse con frecuencia en hojas de cálculo que carecen del control intrínseco necesario para garantizar la integridad de los datos. Por ejemplo:
- Las hojas de cálculo suelen tener una capacidad limitada o nula para limitar la capacidad del usuario de editar los datos.
- Las hojas de cálculo no admiten registros de pistas de auditoría cuando es necesario.
- Cada vez que se añaden o eliminan datos de la hoja de cálculo y se guardan, también se guarda una base de datos completamente nueva; esto significa que alguien que añada datos puede, inadvertida o intencionadamente, cambiar el «código» de la base de datos, y podría pasar desapercibido.
Si se va a desarrollar una solución conforme a la normativa utilizando hojas de cálculo, habrá que desarrollar controles externos para superar estas deficiencias. Aunque existen productos comerciales destinados a proporcionar capacidad de seguimiento de auditoría a las hojas de cálculo tal y como lo realiza eDocuSeed. El usuario debe ser plenamente consciente de las debilidades de las limitaciones de las hojas de cálculo como base de datos cuando se proponga.
Hojas de cálculo como templetes
Un uso muy común de las hojas de cálculo es el desarrollo de soluciones de templetes, donde los datos pueden ser sometidos a una manipulación estándar y los resultados guardados como documento único o incluso exportados a una aplicación. Los templetes se pueden usar, por ejemplo, para tabular y procesar datos de un estudio clínico, o un cálculo de formulario similar basado en los resultados de las pruebas de control de calidad antes del lanzamiento del producto o, el procesamiento de datos de pruebas de laboratorio como son ensayos, contenidos, disoluciones y/o sustancias relacionadas por mencionar algunos.
Al desarrollar tales plantillas, los usuarios y desarrolladores deben comprender y documentar completamente la manipulación requerida. Esto permite establecer y confirmar de manera clara las intenciones de diseño frente a las características estándar lo cual favorece el proceso de validación de datos de entrada, procesamiento y salida de resultados. También es sugerible considerar lo siguiente:
- Se debe verificar que el cálculo sea correcto.
- ¿Se ejecutará la plantilla en una sola estación de trabajo o estará disponible para descargarse desde una sola ubicación? si no,
- ¿Cómo se asegura que todos estén usando la versión correcta? se debe establecer un control de versiones, respaldado por un proceso efectivo de gestión de cambios.
- ¿Cómo se controlará el acceso a los campos de aplicación y datos por parte de los usuarios y desarrolladores? idealmente, todas las celdas que no sean de entrada de datos deberían estar bloqueadas y fuera del alcance de los usuarios.
- ¿Cómo se configurará funcionalmente?
- ¿Existen requisitos de script personalizados al usar el asistente de aplicaciones? una macro es un software personalizado. Incluso cuando se crea mediante la captura del almacén de claves, hay programas en un lenguaje como Visual Basic™ para la aplicación detrás de cada macro.
- ¿Habrá más de un módulo? Las pruebas de integración son apropiadas en tales circunstancias. Para las hojas de cálculo, esto puede implicar enlaces directos de celdas a otras hojas de cálculo. Este enlace puede verse afectado por los cambios y debe abordarse como parte del proceso de control de cambios.
- ¿La entrada de datos será solo a través del teclado? las fuentes de datos externas necesitan configuración, y las hojas de cálculo pueden ser lo suficientemente sofisticadas para manejar entradas inusuales (por ejemplo, una cadena de caracteres que es demasiado larga).
- ¿La salida se guardará en un archivo o solo se imprimirá? los controles de registros electrónicos pueden ser necesarios si el documento se conserva electrónicamente.
- ¿La plantilla maestra se bloqueará y almacenará bajo control de versión en una ubicación separada del uso diario?
- ¿Cómo se aseguran los resultados para proteger la integridad de los datos?
- ¿Deben numerarse automáticamente los formularios cuando se descargan?
- ¿El formulario completo está protegido contra modificaciones adicionales?
- ¿Existe protección para garantizar que el formulario no se elimine ni se reemplace?
¿Existen herramientas en el mercado que mejoren los procesos de control y mantenimiento de libros de Excel?
Como bien sabemos el desafío más grande al que se ha puesto a prueba el sector farmacéutico es el relacionado con la integridad de datos. Derivado de la gran cantidad de datos emitidos día con día en el que se ven impactados documentos y libros de Excel.
Recordemos que la validación de hojas de cálculo es el primer paso. No obstante, muchos libros de Excel que contienen hojas de cálculo utilizados en el sector farmacéutico no cuentan con evidencia de validación lo cual es un riesgo que podrá derivar en un hallazgo critico ante una inspección regulatoria. El segundo paso y el más complejo es adherir las configuraciones necesarias que eleven la integridad de datos y favorezcan la gestión y el mantenimiento de estas con base en GAMP 5 y CFR21.
En la actualidad el trabajo realizado y el menos sugerible es la configuración de control de seguridad y trazabilidad sobre usos de hojas de cálculo con el uso de VBA lo cual es meramente simbólico y a largo mediano y largo plazo poco sostenible en términos de cumplimiento e integridad de datos.
En deappharma tenemos la solución para mitigar tu riesgo por el uso de hojas de cálculo no validadas y con deficiencias en los atributos de control de integridad de datos. Hemos logrado automatizar el proceso y con esto ahorres tiempo, dinero y esfuerzo para la realización de estos servicios de configuración con proveedores externos. Nuestros desarrollos están estructurados bajo un sistema de gestión de calidad trazable y auditable y tales cuales cuentan con el soporte de validación-calificación que garantizan la precisión del alcance del diseño establecido.
Estamos convencidos de que eDocuSeed te ayudara a mitigar el uso de hojas de cálculo y dar ese paso de cumplimiento que necesitas. Conoce sus beneficios y alcance general aquí: ¡Da clic aquí!
“No vivas con el riesgo ¡Mitígalo!” para eso nosotros de ayudamos.
Visita nuestro sitio www.deappharma.com y solicita una demostración y asesoría gratuita.
¡Contáctanos y únete a nuestra comunidad en redes!
21, Abr 2023
Validación/Verificación de procedimientos analíticos enfoque EDQM
La directriz de la ICH sobre «Validación de procedimientos analíticos: Texto y Metodología» (Q2) constituye un análisis de las características de validación que deben tenerse en cuenta durante la validación de un procedimiento analítico (la directriz también se ha adoptado para los veterinarios durante el debate de la VICH). Se dirigen principalmente a la industria farmacéutica
indicando qué datos de validación deben proporcionarse en un expediente de solicitud. Estos datos deben demostrar que las pruebas y los criterios de aceptación propuestos están suficientemente controlados para garantizar una calidad reproducible de los productos en el momento de su comercialización y un control adecuado durante su vida útil (estabilidad).
Dado que las circunstancias en las que trabaja una OMCL son diferentes de las de una empresa farmacéutica – en la mayoría de los casos no se realizan análisis rutinarios, sino que a menudo las respuestas deben en un corto periodo de tiempo, es necesario reconsiderar el grado de validación/verificación antes de realizar un análisis. Por otra parte, en todos casos debe garantizarse que el resultado presentado es fiable. También hay que destacar que los materiales de referencia adecuados son un factor importante tanto para la realización de los los estudios de validación/verificación como en el propio análisis. El uso de preparados de referencia es ampliamente aceptado puede evitar en determinadas circunstancias la consideración de algunas
características de validación, sobre todo en el ámbito de los productos biológicos: esto debe justificarse caso por caso. El OMCL podrá decidir el alcance necesario teniendo en cuenta los factores de riesgo.
El ámbito de aplicación de este documento -dirigido específicamente a los OMCL- es orientar sobre el alcance de la validación/verificación necesaria, en función de diversas circunstancias, es decir, el objetivo del análisis (por ejemplo, detección de incumplimiento), la cantidad de datos de validación ya disponibles (por ejemplo, en caso de transferencia de un método), la experiencia o los datos históricos ya disponibles en el OMCL individual (por ejemplo, recuperación a partir de una matriz compleja; uso rutinario de una valoración estándar incluso si se valoran sustancias diferentes), etc.
Este documento es igualmente aplicable a productos de origen sintético y biológico. No aborda las prácticas habituales de laboratorio: por ejemplo, orientaciones relativas al uso del equipo, calibración, etc.
Este documento es una nota orientativa que ofrece recomendaciones detalladas sobre el de validación/verificación en función de la categoría del procedimiento analítico; cabe señalar que siempre son posibles otros enfoques. Con respecto a la nueva Directiva 2010/63/UE relativa a la utilización ética de los animales para fines científicos y educativos y el convenio europeo sobre protección de los animales vertebrados utilizados para experimentación y Otros Fines Científicos (Consejo de Europa) es necesario prestar especial atención al uso de métodos in vivo como procedimientos analíticos. Debe hacerse todo lo posible para racionalizar y racionalizar y restringir al mínimo necesario la utilización de animales sobre la base de un análisis exhaustivo de la situación.
Cabe destacar que este documento no puede ofrecer asesoramiento detallado para todos los casos posibles en los que se utilicen ensayos in vivo. El propósito de este documento es proporcionar una orientación general. En todos los casos, deberá incluirse una breve descripción y/o justificación del enfoque elegido, incluidos los métodos, deberá describirse en la documentación interna del análisis.
Datos de validación
Deberán justificarse las modificaciones del método validado original. En función de la naturaleza de las modificaciones y del resultado de la evaluación del riesgo, podrán emprenderse actividades de validación o verificación suplementarias. Se aplicarán las mismas definiciones que en el documento ICH.
Categorías de análisis
En este capítulo se definen las diferentes situaciones analíticas (categorías) que pueden darse en un OMCL y las características de validación correspondientes que deben tenerse en cuenta. Consulte la versión actual de la directriz ICH sobre «Validación de procedimientos analíticos»: Text and Methodology» (Q2).
Los estudios formales de validación, de acuerdo con los requisitos de la ICH, deben llevarse a cabo cuando se desarrolle un nuevo método, cuando se utilice un método existente o cuando los datos de validación de un método existente deban completarse.
Según la norma ISO 17025, la validación es necesaria para los métodos no normalizados. .En el contexto de la OMCL, los métodos de farmacopea y los métodos validados de una autorización de comercialización se consideran métodos estándar.
La verificación del método debe realizarse para demostrar que en las condiciones reales de uso en los laboratorios individuales el método (validado) es adecuado (apto para su uso). Esto puede lograrse realizando las pruebas de idoneidad del sistema (por ejemplo, la resolución en un método cromatográfico) el control de la sensibilidad en el umbral de notificación, el control de la integridad de un paso de reacción (por ejemplo, extracción, reacción de hidrólisis) antes de que pueda realizarse la determinación propiamente dicha, verificar la precisión del método, etc. Esto también puede lograrse realizando un ejercicio de transferencia del método, en el laboratorio que ha establecido el método y el OMCL y comparando los resultados para demostrar la equivalencia. En todos los casos, se incluirá una breve nota en la que se expliquen los motivos del método elegido (en función de la complejidad del análisis requerido). La complejidad del análisis requerido-, deberá incluirse en la documentación interna del análisis. Las desviaciones de esta directriz deberán justificarse.
En las tablas 1 a 8 se contemplan varias categorías de análisis:
Tabla 1: Método publicado en la Farmacopea Europea
Los procedimientos analíticos descritos en una monografía se consideran validados. El OMCL debe verificar que todos los materiales de referencia necesarios y que se realizan las pruebas de idoneidad del sistema requeridas. Para las pruebas de sustancias relacionadas, la especificidad para cualquier impureza conocida que no figure en la monografía (por ejemplo, la lista de transparencia Ph Eur). Para las monografías de productos terminados, el OMCL debe verificar que ningún excipiente interfiera en el análisis de la sustancia activa, a menos que se indique contrario en la monografía.
Nota: Para entrar en esta categoría, los procedimientos deben ser descritos en detalle, no por ejemplo como en algunos casos para biológicos donde hay sólo una descripción general del método. Los detalles pueden proceder del informe publicado del estudio colaborativo (por ejemplo, los informes de estudios BSP en Pharmeuropa Bio & Scientific notes)
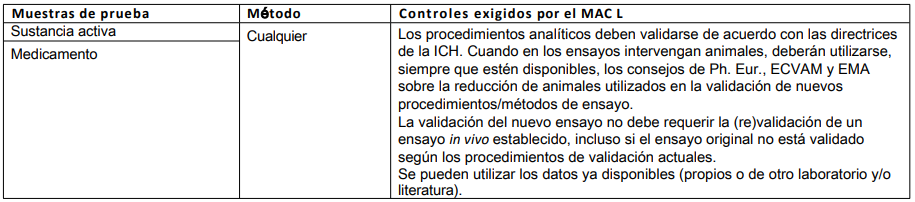
Tabla 2: Método validado de un fabricante (1ª autorización de comercialización)
Los procedimientos analíticos tomados de una autorización de comercialización están totalmente validados por la empresa.
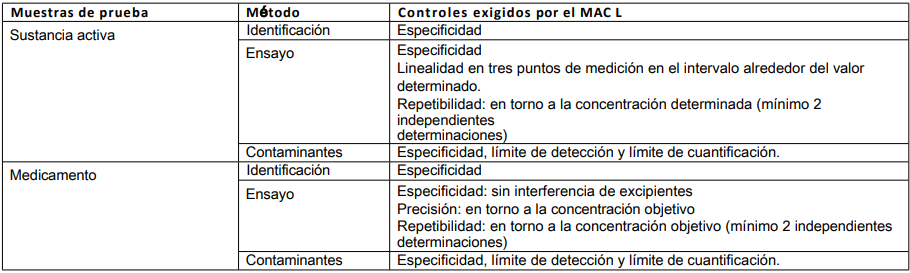
1er MAH = producto fabricado por el MAH que validó el método original utilizado. 2º HAC = producto de un fabricante diferente
para el que no se ha validado específicamente el método original utilizado.
Cuando los métodos procedan de un expediente o expedientes de solicitud antiguos sin datos de validación o con datos insuficientes, se deberá informar a la Autoridad Competente supervisora. Para las características de validación que deben tenerse en cuenta, véanse las tablas 2, 5.
Tabla 3: Método publicado no compendial
Las características de validación a considerar dependerán siempre de la cantidad de datos de validación aportados. Si el método ha sido completamente validado y los datos se han publicado en la bibliografía, véase el cuadro 1. En caso contrario, deberá tenerse en cuenta lo siguiente.
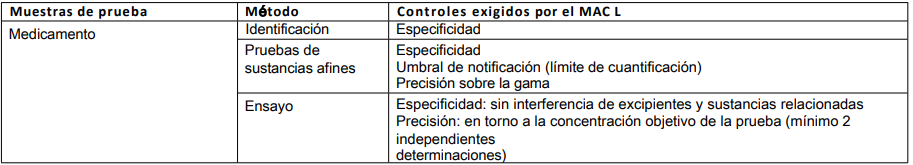
Tabla 4: Método de sustancia activa utilizado para un medicamento
El principal factor a considerar aquí es la influencia de la matriz en el análisis, incluyendo la interferencia de los excipientes.
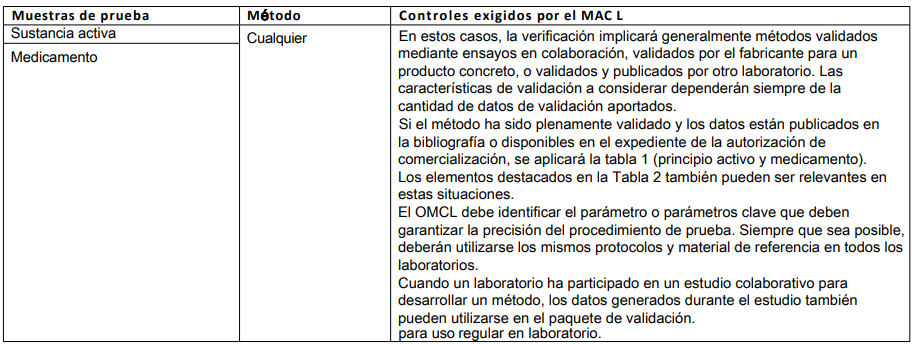
Tabla 5: Métodos validados para reducir, perfeccionar o sustituir el uso de animales (3R)
Tabla 6: Detección de incumplimientos
El cribado de la no conformidad significa que el objetivo del análisis es detectar la posible no conformidad del producto con las especificaciones.
Este tipo de cribado se realizaría cuando se solicita un análisis rápido y/o cuando no se dispone de datos de validación del método. El procedimiento debe documentarse en todos los casos.
Si se detecta un incumplimiento, debe ampliarse el alcance de la validación, por ejemplo, considerando la posibilidad de cambiar a un método bien reconocido (método de compendio o método del expediente MAH).
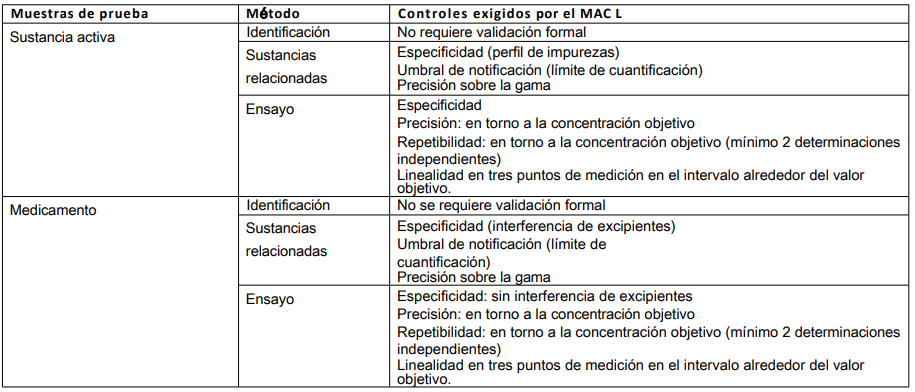
Tabla 7: Detección de productos/contaminantes desconocidos
En estos casos, se carece de información sobre el producto que debe analizarse con respecto a su declaración en la etiqueta (presencia o ausencia de determinadas sustancias) o para aclarar otros aspectos solicitados por la Inspección.
Pruebas a tener en cuenta: identificación, ensayo y, tal vez, pruebas de pureza. El primer paso importante es identificar los principales componentes del producto.
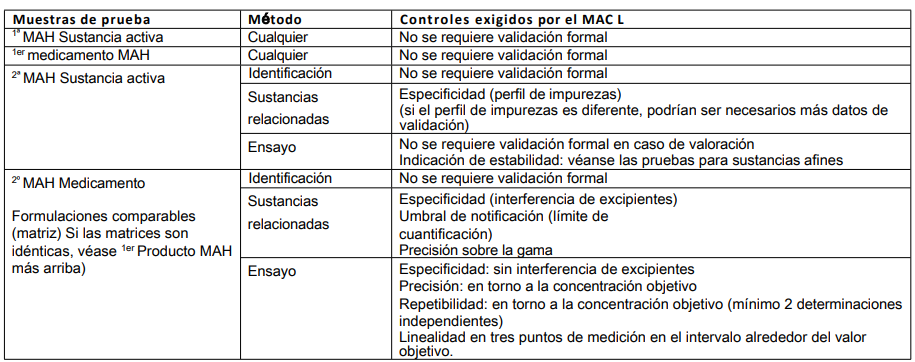
Tabla 8: Desarrollo de un nuevo método
Esto ocurre principalmente cuando un producto se prueba en condiciones de ensayo rutinarias y/o cuando se utiliza un procedimiento analítico interno.

¿Existen herramientas en el mercado que mejoren los procesos administrativos y de laboratorio?
Como bien sabemos el desafío más grande al que se a puesto a prueba el sector farmacéutico es el relacionado con la integridad de datos. Y el proceso de validación de método analíticos no cae fuera de este requerimiento ¡Pero! ¿Por qué es tan complejo este tema? La respuesta se deriva por la gran cantidad de datos y documentos que se emiten día con día en el que se ven impactados documentos y libros de Excel.
Recordemos que la validación de software y libros de Excel es el primer paso. No obstante, muchos libros de Excel utilizados en el sector farmacéutico no cuentan con evidencia de validación lo cual es un riesgo que podrá derivar en un hallazgo critico ante una inspección regulatoria.
El segundo paso y el más complejo es adherir las configuraciones necesarias que eleven la integridad de datos y favorezcan el mantenimiento tanto de software como de Libros de Excel.
Te invitamos a mitigar el riesgo por el uso de sistemas computarizados no validados y con pobres atributos de integridad de datos. Nuestros desarrollos están estructurados bajo un sistema de gestión de calidad trazable y auditable. Tales cuales cuentan con el soporte de validación-calificación documental con base en Gamp 5 y CFR21 parte 11.
Conoce eADMxL el cual es un software que desarrollamos para mitigar el uso de hojas de cálculo para procesos de validación de métodos analíticos. Conoce sus beneficios y alcance general aquí: ¡Da clic aquí!
“No vivas con el riesgo ¡Mitígalo!” para eso nosotros de ayudamos.
Visita nuestro sitio www.deappharma.com y solicita una demostración y asesoría gratuita.
¡Contáctanos! y conoce de que manera te vamos a ayudar.
¡Únete a nuestra comunidad en redes!
Referencias
- General European OMC L Network (GEON) Quality Management Document
- Validation of analitycal procedures :text and methodology Q2
12, Abr 2023
Definiendo los requerimientos de usuario
Una clave muy importante para la validación del software y libros de Excel es una especificación documentada de los requisitos que defina:
– el «uso previsto» del software, libro de Excel o equipo automatizado; y
– la medida en que el fabricante del producto depende de ese software, libro de Excel o equipo para la producción de un producto sanitario de calidad.
El fabricante del producto (usuario) tiene que definir el entorno operativo previsto, incluyendo cualquier configuración de hardware y software necesarias, versiones de software, utilidades, etc. El usuario también debe:
– documentar los requisitos de rendimiento del sistema, calidad, gestión de errores, puesta en marcha, apagado, seguridad, etc;
– identificar cualquier función o característica relacionada con la seguridad, como sensores, alarmas, enclavamientos, pasos lógicos de procesamiento o secuencias de comandos; y
– definir criterios objetivos para determinar el rendimiento aceptable.
La validación debe realizarse de acuerdo con un protocolo documentado, y los resultados de la validación también deben documentarse. Deben documentarse los casos de prueba que pongan a prueba el rendimiento del sistema con respecto a los criterios predeterminados, especialmente para sus parámetros más críticos.
Los casos de prueba deben abordar las condiciones de error y alarma, el arranque, el apagado, todas las funciones de usuario y controles de operador aplicables, posibles errores de operador de valores permitidos y las condiciones de estrés aplicables al uso previsto del equipo. Los casos de prueba deben ejecutarse y los resultados deben registrarse y evaluarse para determinar si los resultados permiten concluir que el programa informático está validado para el uso previsto.
Un fabricante de productos puede llevar a cabo una validación utilizando su propio personal o puede depender de un tercero, como el fabricante del equipo/software tercero, o un consultor. En cualquier caso, el fabricante del producto conserva la responsabilidad última de garantizar que el software del sistema de producción y calidad:
– se valide de acuerdo con un procedimiento escrito para el uso concreto previsto; y
– funcionará según lo previsto en la aplicación elegida.
El fabricante del producto debe disponer de documentación que incluya
– los requisitos de usuario definidos
– el protocolo de validación utilizado
– los criterios de aceptación
– casos de prueba y resultados; y
– un resumen de validación que confirme objetivamente que el software y/o Libros de Excel está validado para el uso previsto.
¿Existen herramientas en el mercado que mejoren los procesos administrativos y de laboratorio?
Como bien sabemos el desafío más grande al que se a puesto a prueba el sector farmacéutico es el relacionado con la integridad de datos. ¡Pero! ¿Por qué es tan complejo este tema? La respuesta se deriva por la gran cantidad de datos y documentos que se emiten día con día en el que se ven impactados documentos y libros de Excel.
Recordemos que la validación de software y libros de Excel es el primer paso. No obstante, muchos libros de Excel utilizados en el sector farmacéutico no cuentan con evidencia de validación lo cual es un riesgo que podrá derivar en un hallazgo critico ante una inspección regulatoria.
El segundo paso y el más complejo es adherir las configuraciones necesarias que eleven la integridad de datos y favorezcan el mantenimiento tanto de software como de Libros de Excel.
Te invitamos a mitigar el riesgo por el uso de sistemas computarizados no validados y con pobres atributos de integridad de datos. Nuestros desarrollos están estructurados bajo un sistema de gestión de calidad trazable y auditable. Tales cuales cuentan con el soporte de validación-calificación documental con base en Gamp 5 y CFR21 parte 11.
Conoce eADMxL el cual es un software que desarrollamos para mitigar el uso de hojas de cálculo para procesos de validación de métodos analíticos. Conoce sus beneficios y alcance general aquí: ¡Da clic aquí!
eDocuSeed es más que un software de gestión documental ya que este es capaz de controlar y mantener libros de Excel por medio de adición de controles necesarios para: acceso, edición, trazabilidad y su mantenimiento. Conoce sus beneficios y alcance general aquí: ¡Da clic aquí!
“No vivas con el riesgo ¡Mitígalo!” para eso nosotros de ayudamos.
Visita nuestro sitio www.deappharma.com y solicita una demostración y asesoría gratuita.
¡Contáctanos! y conoce de que manera te vamos a ayudar.
¡Únete a nuestra comunidad en redes!
24, Nov 2022
Registros electrónicos (Identificadores)
Cuando se gestionan documentos o datos de laboratorio dentro de la industria regulada una función importante dentro del entorno de las buenas prácticas de laboratorio en sistemas computarizados e informatizados es aquel relacionado al control de registros. Visto desde una parte de trazabilidad este identificador es la base de búsqueda de algún documento y/o el comienzo para vincular acciones preestablecidas ya que es de carácter único.
Este articulo estará enfocado hacia el proceso de validación de métodos analíticos, sin embargo el concepto general aplica a diferentes esquemas de software eg, sistemas documentales, datos de laboratorio y hojas de cálculo.
Para comenzar con este articulo compartimos las siguientes definiciones, tales cuales anteceden la esencia de estas líneas.
Como definiciones para el proceso de validación de métodos analíticos establecidas por FDA y USP, respectivamente son;
FDA: “La validación de un método analítico es el proceso por el cual se establece, mediante estudios de laboratorio, que las características de desempeño del método cumplen con los requisitos para las aplicaciones analíticas previstas.”
USP: “La validación de métodos, es el proceso por el cual se demuestra que los procedimientos analíticos son aptos para el uso indicado.”
Como se puede percibir en las definiciones anteriores sobre la validación de métodos analíticos, se haca referencia a la palabra “PROCESO”. La definición de proceso se expresa de la siguiente manera: procesamiento o conjunto de operaciones a que se somete una cosa para elaborarla o transformarla.
Como bien sabemos los procesos de validación de métodos analíticos actualmente son realizados con el uso de hojas de cálculo, donde podemos encontrar varios gaps de integridad de datos. Bajo esta situación el control de registros depende de la clave que se asigna al protocolo de validación correspondiente y por ende el sitio donde se almacena la carpeta física con resultados.
Durante nuestra experiencia hemos observado diferentes situaciones desafortunadas para acceder a la información con procesos de gestión actual, los más representativos son:
- La información no se encuentra en el sitio ubicado destinado en el archivo muerto.
- La información impresa se extrajo y nunca se regresó a su sitio.
- La información impresa se puede destruir y generar un reporte nuevo sin posibilidad de trazar la existencia del primer documento.
Derivado de las situaciones anteriores no solo se observa un impacto sobre los documentos y/o datos de laboratorio. Lo que se impacta de manera esencial es la integridad de los datos y la toma de decisiones.
¿Cómo se impacta la integridad?
La integridad se ve impactada por la falta de control sobre datos y registros. Recordemos que la integridad de datos define que los datos y documentos deben ser perdurables para que estos se mantengan: atribuibles, legibles, contemporáneos y exactos. Por ende, la manera actual de gestionar las validaciones carece de integridad con grandes áreas de oportunidad que se deben atender y solucionar.
¿Que son los registros?
Una parte importante para el control de datos es la estructuración sistemática de registros, tales cuales deben ser ordenados y monitorizados. Pero ¿Qué es un registro?
Un registro es toda la información de un elemento que se almacena en un archivo o tabla de base de datos; por ejemplo, el conjunto de datos debe incluir el nombre del producto, tipo de trabajo, dosis, categoría de método a evaluar, tipo de prueba, referencias de protocolos y vigencias. Así como quien y cuando se creó dicho registro. En algunos casos específicos un registro debe permitir la correlación de este con otros previamente realizados.
Como se mencionó los registros son identificadores únicos tales cuales están estructurados por prefijos, sufijos y números establecidos por algoritmos establecidos. De esta manera se da una secuencia definida sin posibilidad de duplicidad y con una completa trazabilidad mediante pistas de auditoría. Es importante considerar que los registros dentro de los sistemas computarizados e informatizados deben ser ordenados e identificados de manera secuencial para una búsqueda eficiente y un acceso rápido.
¿Nosotros como lo hacemos?
A continuación, te mostramos la secuencia de pasos que realiza nuestro sistema estrella para la creación, control y acceso a registros de validación de métodos analíticos. No obstante, te invitamos a revisar nuestros otros desarrollos, tales cuales cuentan también con estas funciones.
Vallamos entonces.
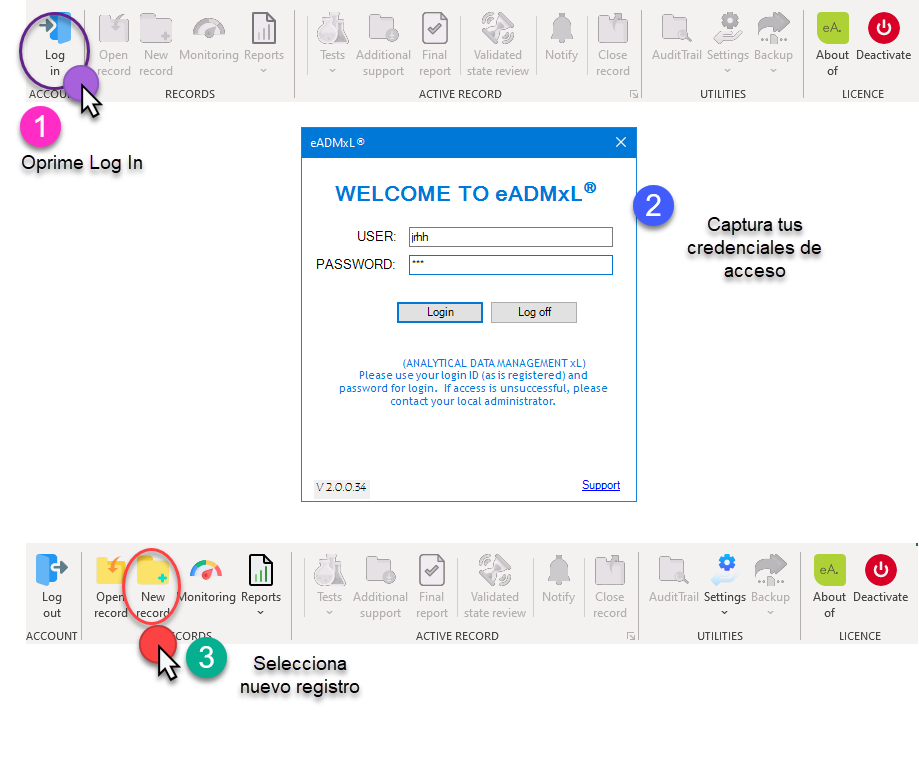
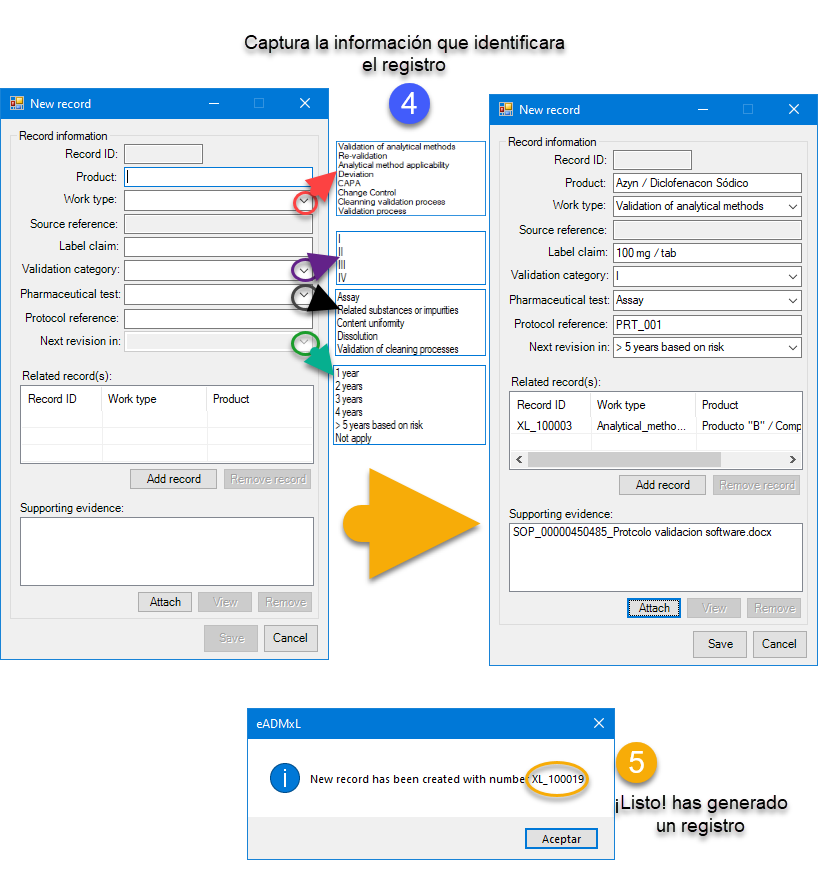
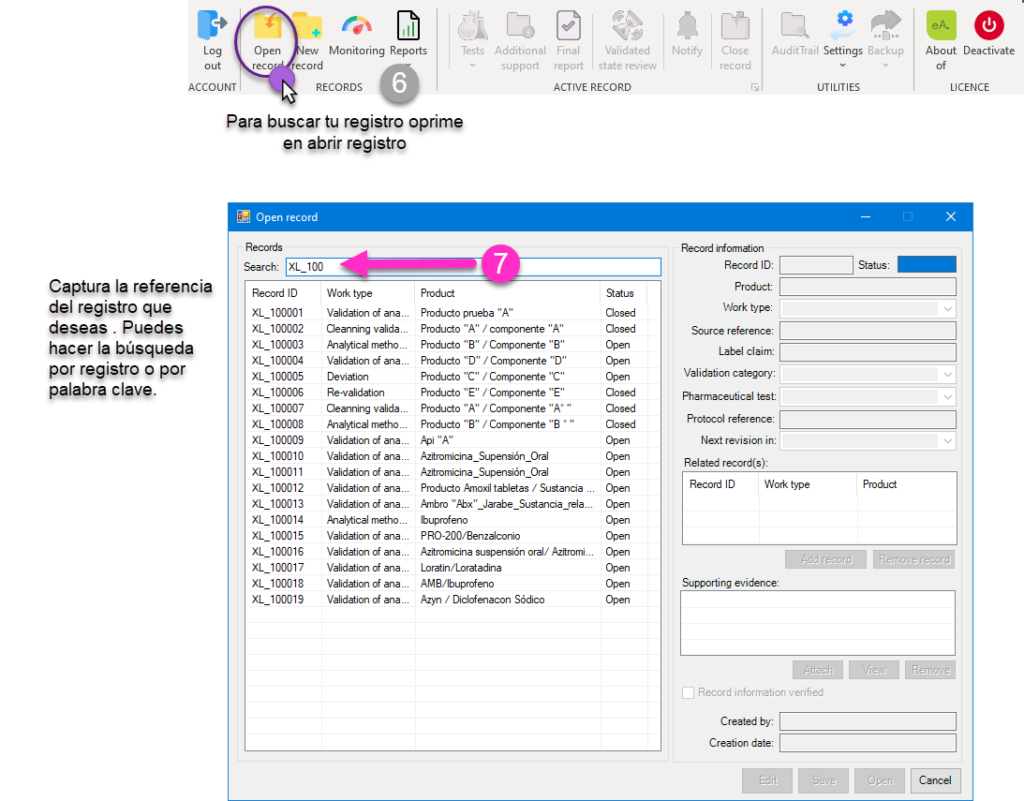
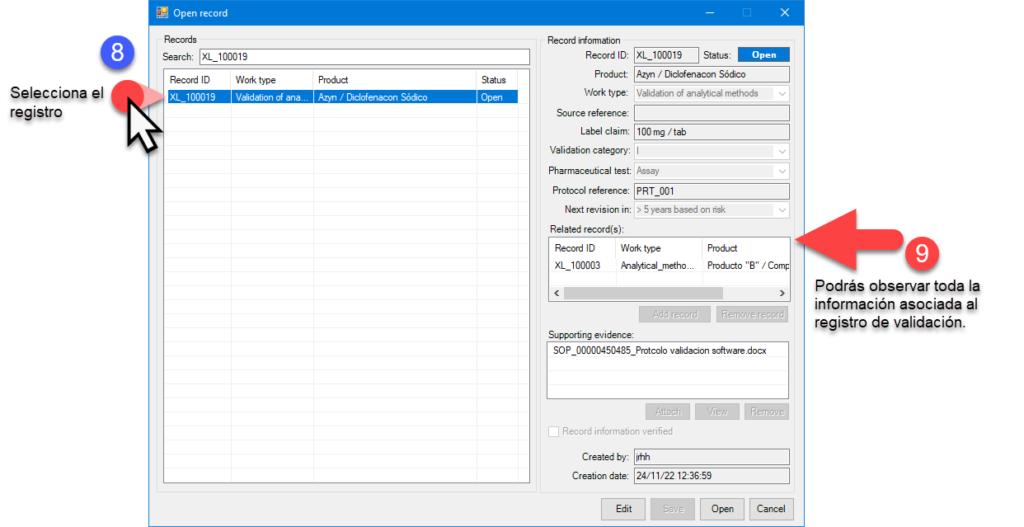
En la actualidad hay muchos gaps relacionados al acceso y control de sistemas computarizados (se incluyen hojas de cálculo). Es por esto que en deappharma desarrollamos herramientas que potencian tu cumplimiento de manera fácil y lógica. Te invitamos a conocer y solicitar tu DEMO y/o implementación de licencia gratuita de nuestro sistema estrella eADMxL para realizar el proceso de validación de métodos analíticos. También te invitamos a explorar nuestros otros desarrollos y visualizar en que proceso te podemos ayudar.
El cambio está en tus manos. Nosotros de ayudamos a potenciar tu cumplimiento.
Referencias:
- FDA draft guidance – Analytical Procedures and Methods Validation
- USP 31 –NF 26, Capítulo General Validación de métodos farmacopéicos
- Villareal de la Garza Sonia, Introducción a la computación. Cap 8 Ed. McGrawHill , México , 2007
¿Necesitas ayuda?
¡Contáctanos! y conoce de que manera te vamos a ayudar.
¡Únete a nuestra comunidad en redes!
15, Sep 2022
Firmas electrónicas (Digitales)
Firmas electrónicas
Las regulaciones del FDA Título 21 CFR Parte 11 establece los criterios sobre los cuales el FDA considera los documentos electrónicos, firmas electrónicas y firmas escritas a mano realizadas a los documentos electrónicos para ser confiables, fiables y generalmente equivalentes a los registros de papel y firmas escritas a mano ejecutadas en papel. Los requisitos generales que establece la FDA son;
(a) Cada firma electrónica deberá ser única para un individuo y no deberá ser reutilizada por, o reasignada a, nadie más.
(b) Antes de que una organización establezca, asigne, certifique o sancione de otro modo la firma electrónica de un individuo, o cualquier elemento de dicha firma electrónica, la organización deberá verificar la identidad del individuo.
(c) Las organizaciones que utilicen firmas electrónicas deberán, antes o en el momento de su utilización, certificar de manera interna que estas están destinadas a ser el equivalente legalmente vinculante de las firmas manuscritas tradicionales.
Componentes y controles de la firma electrónica.
(a) Las firmas electrónicas que no se basen en la biometría deberán
(1) Emplear al menos dos componentes de identificación distintos, como un código de identificación y una contraseña.
Cuando un individuo ejecuta una serie de firmas durante un período único y continuo de acceso controlado al sistema, la primera firma deberá ser ejecutada usando todos los componentes de la firma electrónica; las firmas subsecuentes deberán ser ejecutadas usando por lo menos un componente de la firma electrónica que es solamente ejecutable por, y diseñado para ser usado solamente por, el individuo.
Cuando una persona ejecute una o más firmas que no se realicen durante un período único y continuo de acceso controlado al sistema, cada firma se ejecutará utilizando todos los componentes de la firma electrónica.
(2) Ser utilizadas únicamente por sus auténticos propietarios; y
(3) Ser administrada y ejecutada para asegurar que el intento de uso de la firma electrónica de un individuo por cualquiera que no sea su genuino propietario requiera la colaboración de dos o más individuos.
(b) Las firmas electrónicas basadas en la biometría se diseñarán para garantizar que no puedan ser utilizadas por nadie más que sus auténticos propietarios.
Controles de los códigos de identificación/contraseñas.
Las personas que utilicen firmas electrónicas basadas en el uso de códigos de identificación en combinación con contraseñas deberán emplear controles para garantizar su seguridad e integridad. Dichos controles incluirán:
(a) Mantener la singularidad de cada código de identificación y contraseña combinados, de manera que no haya dos personas que tengan la misma combinación de código de identificación y contraseña.
(b) Garantizar que las emisiones de códigos de identificación y contraseñas se comprueben, retiren o revisen periódicamente (por ejemplo, para cubrir eventos como el envejecimiento de las contraseñas).
Expertos en software comercial y servicios de validación de sistemas computarizados e informatizados
No olvides que en caso de ser necesario, puedes contactarnos para mejorar el proceso de laboratorio y/o administrativo de tu organización a través software 100% confiable y seguro.
Recuerda que en deappharma contamos con software que te ayudara a administrar y controlar procesos especializados. Somos expertos en la materia, por lo que procesos complejos no representa un reto para nosotros, debido a que tenemos en mente los aspectos relevantes de cumplimiento.
¡Contáctanos! y conoce de que manera te vamos a ayudar.
¡Únete a nuestra comunidad en redes!
2, May 2022
Características de un buen SRS (Software Requirements Specification) especificación de requerimientos de software
La especificación de requisitos de software (SRS) es una descripción completa del comportamiento del sistema que se va a desarrollar. Incluye un conjunto de casos de uso que describe todas las interacciones que tendrán los usuarios con el software. Los casos de uso también son conocidos como requisitos funcionales. Además de los casos de uso, la ERS también contiene requisitos no funcionales (complementarios). Los requisitos no funcionales son requisitos que imponen restricciones en el diseño o la implementación, como, por ejemplo, restricciones en el diseño o estándares de calidad.
Está dirigida tanto al cliente como al equipo de desarrollo. El lenguaje utilizado para su redacción debe ser informal, de forma que sea fácilmente comprensible para todas las partes involucradas en el desarrollo.
Las características de un buen SRS son:
- Sin ambigüedades
- Completa
- Verificable
- Coherente
- Modificable
- Trazable
- Utilizable durante la fase explotación y mantenimiento
Cada característica se aborda con mayor detalle a continuación:
Sin ambigüedad. Una SRS es inequívoca si- y solo si- cada requisito en ella tiene una única interpretación.
- Como mínimo, esto requiere que cada característica del producto final se describa utilizando un único termino.
- En los casos en los que un término utilizado en un contexto particular pueda tener múltiples significados, el termino debe incluirse en un glosario donde se especifique su significado.
Completa. Un SRS es completo si posee las siguientes cualidades:
- Inclusión de todos los requisitos significativo, ya sean relativos a la funcionalidad, el rendimiento, las limitaciones de diseño, los atributos o las interrelaciones externas.
- Definición de las respuestas de software a todas las clases realizables de datos de entrada en todas las clases realizables de situaciones. Obsérvese que es importante especificar la respuesta a los valores de entrada válidos y no validados.
- Conformidad con cualquier norma del SRS que le sea aplicable. Si una sección particular de la norma no es aplicable, el SRS debe incluir el número de sección y una explicación de porque no es aplicable.
Verificable. Un SRS es verificable si, y solo si, todos los requisitos establecidos en el son verificables. Un requisito es verificable si y solo si existe algún proceso finito y rentable con el que una persona o máquina pueda comprobar que el producto de software cumple con el requisito.
- Ejemplos de requisitos no verificables, estas son declaraciones como: a)El producto debe funcionar bien, o el producto debe tener una buena interfaz humana. Estos requisitos no pueden verificarse porque es imposible definir los términos bueno o bien. b)El programa nunca debe entrar en un bucle infinito. Este requisito no es verificable porque la comprobación de esta cualidad es teóricamente imposible.
Coherente. Un buen SRS es consistente si y solo si ningún conjunto de requisitos individuales descritos en ella entra en conflicto. Hay tres tipos de conflictos principales en un SRS:
- Dos o más requisitos pueden describir el mismo objeto del mundo real, pero utilizar termino diferentes para ese objeto. Por ejemplo, la solicitud de una entrada de usuarios por parte de un programa puede denominarse “prompt” en un requisito y “cue” en otro.
- Las características especificadas de los objetos del mundo real pueden entrar en conflicto. Por ejemplo: 1)El formato de un informe de salida puede describirse en un requisito como «tabular” pero en otro como “textual” 2) Un requisito puede establecer que todas las luces deben ser verdes mientras que otro establece que todas las luces deben ser azules. 3) Puede haber un conflicto lógico o temporal entre dos acciones específicas. Por ejemplo; a) Un requisito podría especificar que el programa sumará dos entradas y otro especificar que el programa las multiplicará. b) Un requisito puede establecer que A debe seguir a B, mientras que otros requieren que A y B ocurran simultáneamente.
Modificable. Un SRS es modificable si su estructura y estilo son tales que cualquier cambio necesario en el requisito puede realizarse de forma fácil, completa y coherente. Los cambios en un SRS generalmente requieren:
- Tener una organización coherente y fácil de usar, con un índice y referencias cruzada explicitas.
- No ser redundante; es decir, el mismo requisito no debe aparecer en otras partes en el SRS.
Trazable. Una SRS es trazable si el origen de cada uno de sus requisitos es claro y se facilita la referencia de cada requisito de la futura documentación de desarrollo o mejora. Se recomiendan dos tipos de trazabilidad:
- La trazabilidad hacia atrás (es decir, hacia etapas anteriores de desarrollo) depende de que cada requisito haga referencia explícita a su fuente en documentos anteriores.
- La trazabilidad hacia adelante (es decir, hacia todos los documentos generados por el SRS) depende que cada requisito del SRS tenga un nombre o número de referencia único.
Utilizable durante la fase de operación y mantenimiento. El SRS debe atender las necesidades de la fase de operación y mantenimiento, incluyendo la eventual sustitución del software.
- El mantenimiento suele ser realizado por personal no relacionado con el desarrollo original. Los cambios locales (correcciones) pueden aplicarse mediante un código. Si embargo, para los cambios de mayor alcance, la documentación sobre el diseño y los requisitos es esencial. Esto implica dos acciones:
- El SRS debe ser modificable
- El SRS debe contener un registro de todas las disposiciones especiales que aplican a los componentes individuales, tales como:
- Su criticidad
- Su relación con las necesidades sólo temporales.
- Su origen
- Este tipo de conocimiento se da por sentado en la organización de desarrollo, pero a menudo falta en la organización de mantenimiento. Si no se entiende la razón o el origen de una función, a menudo es imposible realizar un mantenimiento adecuado del software.
No olvides que todas nuestras soluciones son liberadas bajo un sistema de calidad robusto. En caso de ser necesario, puedes contactarnos para mejorar el proceso de laboratorio y administrativo de tu organización a través software 100% confiable y seguro. Recuerda que en deappharma contamos con una suite de herramientas que te ayudaran a mejorar tus procesos con un enfoque en integridad de datos. Somos expertos en la materia, por lo que procesos complejos no representa un reto para nosotros, debido a que tenemos en mente los aspectos relevantes de cumplimiento.
Referencias
-
FDA, Guidance for the Content of Premarket Submissions for Software Contained in Medical Devices
-
GAMP 5 Guide: Compliant GxP Computerized Systems
-
Guía del IEEE sobre especificaciones de requisitos de software ANSI/IEEE std 1984.
¡Contáctanos! y conoce de que manera te vamos a ayudar.
¡Únete a nuestra comunidad en redes!
11, Feb 2022
Audit Trail para asegurar y elevar el proceso de Integridad de datos
Este término regulatorio e informático se refiere a la auditoría, detección continua y exhaustiva de los datos. Por lo que es relevante contar con este proceso o función en cada uno de nuestros sistemas informatizados. El objetivo, es trazar la vida de un dato de manera perdurable.
La industria farmacéutica, dispositivos médicos y almacenes utilizan sistemas informatizados dentro de sus procesos. Por este hecho, es relevante el control y trazabilidad de datos. Para abordar el tema y conocer el alcance e importancia de la función de pistas de auditoria te invitamos a leer lo siguiente.
¿Qué es un “pista de auditoría”?
La «Pista de auditoría» o también conocida como «Audit Trail». Significa un registro electrónico seguro, generado por computadora con sellos de tiempo que permiten la reconstrucción del curso de los eventos relacionados con la creación, modificación y/o eliminación de un registro electrónico. Por ejemplo, la pista de auditoría para una ejecución de cálculos analíticos con la utilización de Excel como software de cálculo, debe incluir como mínimo lo siguiente: nombre de usuario, la fecha/hora de la ejecución, los cambios realizados sobre un valor y los detalles justificados de dicho cambio, según apliquen. ¡Pero no solo eso! Las pistas de auditoria son un aparte esencial de los sistemas computarizados. Es por esto, que los reportes de auditorita deben ser robustos y seguros.
Retomemos el ejemplo de uso de la hoja de Excel para obtención de resultados analíticos. En la actualidad la industria configura la obtención de pistas de auditoría sobre los mismos libros de Excel. Esta pista «LOG» regularmente la establecen dentro del mismo libro de manera anidada como una pestaña (hoja) de cálculo. ¡Esta práctica, es poco sostenible y segura! ya que la trazabilidad la dejas en manos de Excel y de la vulnerabilidad de código realizado con VBA.
¿Existe riesgo de perder la trazabilidad en mis libros de Excel por usar configuración VBA?
La respuesta es ¡Si! y tu impacto será alto porque no tendrás la manera de trazar la información.
Los riesgos los podemos clasificar en dos escenarios posibles:
- Daño en el archivo de Excel que no permita acceder a la información.
- Alteración y vulnerabilidad de la programación en VBA (Hasta videos en youtube te enseñan a desbloquear código relizadoo con VBA. ¡Ojo con esto!)
¿Quién debe revisar los registros de auditoría?
La revisión de la pista de auditoría es similar a la evaluación de las tachaduras en papel al revisar los datos. Personal responsable de la revisión de registros bajo cGMP debe revisar las pistas de auditoría que capturan los cambios en datos asociados con el registro a medida que revisan el resto del registro. Para ejemplo, todos los registros de producción y control, que incluyen registros de auditoría, deben revisarse y aprobado por la unidad de calidad. Las regulaciones brindan flexibilidad para tener algunas actividades revisadas por una persona que supervisa o verifica directamente la información.
FDA recomienda un enfoque de sistema de calidad para implementar la supervisión y revisión de cGMP registros.
¿Con qué frecuencia se deben revisar los registros de auditoría?
Si la frecuencia de revisión de los datos se especifica en las regulaciones de cGMP, adhiérase a esa frecuencia para la revisión de la pista de auditoría. Por ejemplo, requiere revisión después de cada paso significativo en fabricación, procesamiento, empaque o almacenamiento, y requiere la revisión de datos antes del lote liberar. En estos casos, aplicaría la misma frecuencia de revisión para la pista de auditoría.
Si la frecuencia de revisión de los datos no está especificada en las regulaciones de cGMP, debe determinar la frecuencia de revisión de la pista de auditoría utilizando el conocimiento de sus procesos y la evaluación de riesgos.
La evaluación de riesgos debe incluir la evaluación de la criticidad de los datos, los mecanismos de control e impacto en la calidad del producto. Los riesgos para los datos incluyen, entre otros, la posibilidad de que se eliminen modifique o excluyan sin autorización o sin detección.
Su enfoque para la revisión de la pista de auditoría y la frecuencia con la que la realiza deben garantizar que se cumplen los requisitos de cGMP, se implementan los controles apropiados y la confiabilidad de la revisión.
¿Puedo aumentar la trazabilidad de mis libros de Excel sin necesidad de usar VBA?
¡Si! y todo de manera virtual con el suso de nuestro complemento COM. Esto garantiza que tus datos sean trazados de manera perdurable.
En deappharma hemos desarrollado eDocuSeed una herramienta validada y especializada para el mantenimiento, gestión y trazabilidad de libros de Excel. ¡Sin duda te ayudaremos a mitigar tu riesgo! Puedes contactarnos para mejorar el proceso administrativo de tu organización a través software 100% confiable y seguro.
Referencias
- Data Integrity and Compliance With Drug CGMP Questions and Answers Guidance for Industry
- Guía para el enfoque de sistemas de calidad de la industria para las regulaciones CGMP farmacéuticas.
¡Contáctanos! y conoce de que manera te vamos a ayudar.
¡Únete a nuestra comunidad en redes!
24, Ene 2022
Detalles del Enfoque – Alcance de la Parte 11 de CFR
Detalles del Enfoque – Alcance de la Parte 11
Interpretación estrecha del alcance
Entendemos que existe cierta confusión sobre el alcance de la parte 11. Algunos han entendido que el alcance de la parte 11 es muy amplio. Creemos que algunas de esas amplias interpretaciones podrían conducir a controles y costos innecesarios y podrían desalentar la innovación y los avances tecnológicos sin brindar un beneficio adicional a la salud pública. Como resultado, queremos aclarar que la Agencia tiene la intención de interpretar el alcance de la parte 11 de manera restringida.
Según la interpretación restringida del alcance de la parte 11, con respecto a los registros que deben mantenerse según las reglas predicadas o presentarse a la FDA, cuando las personas optan por utilizar registros en formato electrónico en lugar de papel, se aplicaría la parte 11. Por otro lado, cuando las personas usan computadoras para generar impresiones en papel de registros electrónicos, y esos registros en papel cumplen con todos los requisitos de las reglas predicadas aplicables y las personas confían en los registros en papel para realizar sus actividades reguladas, la FDA generalmente no condicionara a las personas para «usar registros electrónicos en lugar de registros en papel». En estos casos, el uso de sistemas informáticos en la generación de registros en papel no activaría la parte 11.

Definición de registros de la Parte 11
Bajo esta interpretación estrecha, la FDA considera que la parte 11 es aplicable a los siguientes registros o firmas en formato electrónico (registros o firmas de la parte 11):
- Registros que deben mantenerse según los requisitos de la regla establecida y que se mantienen en formato electrónico en lugar del formato en papel. Por otro lado, los registros (y cualquier firma asociada) que no se requiera conservar según las reglas establecida, pero que, sin embargo, se mantienen en formato electrónico, no son registros de la parte 11.
Le recomendamos que determine, en función de las reglas establecidas, si los registros específicos son registros de la parte 11. Le recomendamos que documente tales decisiones.
- Registros que deben mantenerse según las reglas establecidas, que se mantienen en formato electrónico además del formato en papel, y en los que se confía para realizar actividades reguladas.
En algunos casos, las prácticas comerciales reales pueden dictar si está utilizando registros electrónicos en lugar de registros en papel. Por ejemplo, si se requiere que se mantenga un registro bajo una regla predicada y usted usa una computadora para generar una copia impresa en papel de los registros electrónicos, pero aun así depende del registro electrónico para realizar actividades reguladas, la Agencia puede considerar que usted está utilizando el registro electrónico en lugar del registro en papel. Es decir, la Agencia puede tener en cuenta sus prácticas comerciales para determinar si se aplica la parte 11.
En consecuencia, recomendamos que, para cada registro que deba mantenerse según las reglas establecidas, determine de antemano si planea confiar en el registro electrónico o en el registro en papel para realizar actividades reguladas. Recomendamos que documente esta decisión (por ejemplo, en un Procedimiento operativo estándar (SOP) o documento de especificación).
Registros enviados a la FDA, conforme a reglas establecidas (incluso si dichos registros no están específicamente identificados en las reglamentaciones de la Agencia) en formato electrónico. Sin embargo, un registro que no se presenta en sí mismo, pero que se utiliza para generar una presentación, no es un registro de la Parte 11 a menos que se requiera que se mantenga bajo una regla establecida y se mantenga en formato electrónico.
Firmas electrónicas que pretenden ser el equivalente de firmas manuscritas, iniciales y otras firmas generales requeridas por reglas establecidas. Las firmas de la Parte 11 incluyen firmas electrónicas que se utilizan, por ejemplo, para documentar el hecho de que ciertos eventos o acciones ocurrieron de acuerdo con la regla establecida (por ejemplo, aprobado, revisado y verificado).
Enfoque de los requisitos específicos de la Parte 11
Validación
La Agencia tiene la intención de ejercer la discreción de cumplimiento con respecto a los requisitos específicos de la parte 11 para la validación de sistemas computarizados y los requisitos correspondientes. Aunque las personas aún deben cumplir con todos los requisitos de las reglas establecidas aplicables para la validación, esta guía no debe interpretarse como una imposición de requisitos adicionales para la validación.
Sugerimos que su decisión de validar los sistemas computarizados y el alcance de la validación tengan en cuenta el impacto que los sistemas tienen en su capacidad para cumplir con los requisitos de las reglas establecidas. También debe considerar el impacto que esos sistemas podrían tener en la precisión, confiabilidad, integridad, disponibilidad y autenticidad de los registros y firmas requeridos. Incluso si no existe un requisito de regla de establecido para validar un sistema, en algunos casos puede ser importante validar el sistema.
Recomendamos que base su enfoque en una evaluación de riesgos justificada y documentada y una determinación del potencial del sistema para afectar la calidad y seguridad del producto y la integridad del registro. Por ejemplo, la validación no sería importante para un procesador de texto que se usa solo para generar SOP.
Pista de auditoría
La Agencia tiene la intención de ejercer la discreción de cumplimiento con respecto a los requisitos específicos de la parte 11 relacionados con las pistas de auditoría generadas por computadora y con sello de tiempo y cualquier requisito correspondiente. Las personas aún deben cumplir con todos los requisitos de reglas establecidas aplicables relacionados con la documentación de, por ejemplo, fecha, hora o secuencia de eventos, así como cualquier requisito para garantizar que los cambios en los registros no oscurezcan entradas previas.
Incluso si no existen requisitos de reglas establecidas para documentar, por ejemplo, la fecha, la hora o la secuencia de eventos en una instancia particular, puede ser importante contar con pistas de auditoría u otras medidas de seguridad físicas, lógicas o de procedimiento para garantizar la confiabilidad y confiabilidad de los registros. Le recomendamos que base su decisión de aplicar pistas de auditoría u otras medidas apropiadas, en la necesidad de cumplir con los requisitos de la regla establecida, una evaluación de riesgos justificada y documentada, y una determinación del efecto potencial sobre la calidad y seguridad del producto y la integridad del registro. Le sugerimos que aplique los controles apropiados basados en dicha evaluación. Los registros de auditoría pueden ser particularmente apropiados cuando se espera que los usuarios creen, modifiquen o eliminen registros regulados durante el funcionamiento normal.
Sistemas heredados
La Agencia tiene la intención de ejercer la discreción de ejecución con respecto a todos los requisitos de la parte 11 para los sistemas que de otro modo estaban operativos antes del 20 de agosto de 1997, la fecha de vigencia de la parte 11, bajo las circunstancias especificadas a continuación.
Esto significa que la Agencia no tiene la intención de tomar medidas de cumplimiento para imponer el cumplimiento de los requisitos de la parte 11 si se cumplen todos los siguientes criterios para un sistema específico:
- El sistema estaba operativo antes de la fecha de vigencia.
- El sistema cumplió con todos los requisitos de reglas establecidas aplicables antes de la fecha de vigencia.
- Actualmente, el sistema cumple con todos los requisitos de reglas establecidas aplicables.
- Tiene evidencia documentada y justificación de que el sistema es apto para el uso previsto (lo que incluye tener un nivel aceptable de seguridad e integridad de registros, si corresponde).
Si un sistema ha cambiado desde el 20 de agosto de 1997, y si los cambios impiden que el sistema cumpla con los requisitos de la regla establecidad, los controles de la Parte 11 deben aplicarse a los registros y firmas de la Parte 11 de conformidad con la política de cumplimiento expresada en esta guía.
Copias de Registros
La Agencia tiene la intención de ejercer la discreción de ejecución con respecto a los requisitos específicos de la parte 11 para generar copias de registros y cualquier requisito correspondiente. Debe proporcionar a un investigador un acceso razonable y útil a los registros durante una inspección. Todos los registros en su poder están sujetos a inspección de acuerdo con las reglas establecidas.
Recomendamos que proporcione copias de registros electrónicos por:
- Producir copias de registros mantenidos en formatos portátiles comunes cuando los registros se mantienen en estos formatos
- Usar métodos de conversión o exportación automatizados establecidos, cuando estén disponibles, para hacer copias en un formato más común (los ejemplos de dichos formatos incluyen, entre otros, PDF, XML o SGML)
En cada caso, recomendamos que el proceso de copiado produzca copias que conserven el contenido y el significado del registro. Si tiene la capacidad de buscar, ordenar o buscar tendencias en los registros de la parte 11, las copias entregadas a la Agencia deben proporcionar la misma capacidad si es razonable y técnicamente factible. Debe permitir la inspección, revisión y copia de registros en un formato legible en su sitio utilizando su hardware y siguiendo sus procedimientos y técnicas establecidos para acceder a los registros.
Retención de registros
La Agencia tiene la intención de ejercer la discreción de ejecución con respecto a los requisitos de la parte 11 para la protección de registros para permitir su recuperación precisa y rápida durante todo el período de retención de registros y cualquier requisito correspondiente. Las personas aún deben cumplir con todos los requisitos de reglas establecidas aplicables para la retención y disponibilidad de registros.
Sugerimos que su decisión sobre cómo mantener los registros se base en los requisitos de las reglas predicadas y que base su decisión en una evaluación de riesgos justificada y documentada y en una determinación del valor de los registros a lo largo del tiempo.
FDA no tiene la intención de objetar si usted decide archivar los registros requeridos en formato electrónico en medios no electrónicos como microfilm, microficha y papel, o en un formato de archivo electrónico estándar (ejemplos de tales formatos incluyen, entre otros, PDF, XML o SGML). Las personas aún deben cumplir con todos los requisitos de las reglas establecidas, y los propios registros y cualquier copia de los registros requeridos deben conservar su contenido y significado. Siempre que los requisitos de la regla establecida se cumplan por completo y el contenido y el significado de los registros se conserven y archiven, puede eliminar la versión electrónica de los registros. Además, los componentes de firmas y registros en papel y electrónicos pueden coexistir (es decir, una situación híbrida) siempre que se cumplan los requisitos de la regla establecida y se conserve el contenido y el significado de esos registros.
No olvides que en caso de ser necesario, puedes contactarnos para mejorar el proceso de laboratorio y administrativo de tu organización a través software 100% confiable y seguro. Recuerda que en deappharma somos expertos en la materia, por lo que procesos complejos no representa un reto para nosotros, debido a que tenemos en mente los aspectos relevantes de cumplimiento.
REFERENCES
- Glossary of Computerized System and Software Development Terminology (Division of Field Investigations, Office of Regional Operations, Office of Regulatory Affairs, FDA 1995)
- General Principles of Software Validation; Final Guidance for Industry and FDA Staff (FDA, Center for Devices and Radiological Health, Center for Biologics Evaluation and Research, 2002)
- Guidance for Industry, FDA Reviewers, and Compliance on Off-The-Shelf Software Use in Medical Devices (FDA, Center for Devices and Radiological Health, 1999)
- Pharmaceutical CGMPs for the 21st Century: A Risk-Based Approach; A Science and Risk-Based Approach to Product Quality Regulation Incorporating an Integrated Quality Systems ApproachExternal Link Disclaimer (FDA 2002)
¡Contáctanos! y conoce de que manera te vamos a ayudar.
¡Únete a nuestra comunidad en redes!