Importancia de la validación.
Validación se puede definir como “encontrar o evaluar la verdad de algo”.
Cuando un método analítico es usado para generar resultados acerca de las características de la sustancia de interés (eg., fármaco o medicamento), es vital que los resultados sean confiables, ya que estos serán usados como base para la toma de decisiones en relación, con la definición de la forma de administración del fármaco o medicamento al paciente.
Un estudio de validación, el cual deriva en un método analítico tiene como finalidad el asegurar que los resultados obtenidos sean fidedignos (ALCOA), siempre que estos sean obtenidos dentro del proceso analítico.
¿Pero en donde o porque radica la importancia del proceso de validación?
La importancia radica en que cada día, se realizan en el mundo un alto número de análisis, relacionados al monitoreo de compuestos orgánicos, estas mediciones son utilizadas en algunas situaciones para la toma de decisiones, en áreas como son; control de calidad (liberación), manufactura (evaluación de procesos), áreas analíticas (estudios de estabilidad), proyectos de transferencia analítica.
El límite de Cuantificación “Concepto”
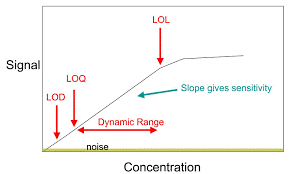
Límite de cuantificación, es la habilidad de un método para detectar y cuantificar la cantidad mínima de un analítico en una muestra, cual puede ser detectada con precisión y exactitud.
El límite de cuantificación es un parámetro de cuantificación de ensayo, para los niveles bajos de los compuestos en las matrices farmacéuticas, y es usada particularmente para la determinación de impurezas y/o productos de degradación.
Planteamiento de la prueba.
Se preparan 6 muestras de placebo adicionadas con el componente principal Api (polvo) (Active Pharmaceutical Ingredient) es lo recomendable. Ya que de esta manera podrás retar la esencia de la preparación analítica. Es recurrente contar con una cantidad limitada de impureza, por lo que, se podrá plantear la prueba con un estándar ya disuelto, para posteriormente ser adicionado a las soluciones placebo (n=6) y de esta manera optimizar el uso.
En caso de no contar con el placebo o matriz farmacéutica se podrán preparar 6 muestras independientes de producto terminado caduco o producto que es sometido a pruebas de estrés controlada. La concentración analítica de la muestra estará determinada en relación al 50% ó 0.05% respecto a la concentración establecida, es decir, respecto al límite de sustancias relacionadas, productos de degradación conocidos y/o desconocidos establecidos en tu método de análisis. El restante porcentual será completado con producto terminado, en los que el cálculo de recobro se vera ajustado por la diferencia que habrá entre el nivel evaluado y el blanco (muestra enriquecida) para ajustar la señal de trabajo.
Estadística y parámetros a evaluar (primarios).
- Promedio individual
- Promedio de n=6
- Desviación estándar de n=6
- Mínimo de n=6
- Máximo de n=6
- Intervalos de confianza del valor de “µ”
- T critico
- T estadístico
- Intervalo de confianza de ““µ” %
- P-value
Criterios de aceptación.
La evaluación deberá mostrar las señales del pico de interés diferenciadas de la señal ruido:
- Señal ruido del pico de interés mínimo de 10.
- Los recobros de las 6 muestras deben cumplir con el recobro especificado.
- Debe mostrarse homocedasticidad en los resultados globales n=6, p-value mayor al 0.05
- Los intervalos de confianza deben incluir el valor del 100%.
- El coeficiente de valoración debe ser “<= “al establecido 5, 10, 15 ó 20 % según aplique el nivel % del límite evaluado.
Obtención de la concentración a evaluar.
Las principales formas de obtención del parámetro son:
1.-Basado en evaluación visual: la evaluación visual puede usarse para métodos no instrumentales, pero también puede usarse con métodos instrumentales. el límite de cuantificación se determina mediante el análisis de muestras con concentraciones de analito y estableciendo el nivel mínimo al cual el analito puede ser detectado de manera confiable.
2.-Basado en señal a ruido: este enfoque solo se puede aplicar a los procedimientos analíticos que muestran una línea de base ruido. La determinación de la relación señal / ruido se realiza comparando señales de muestras con bajas concentraciones conocidas de analito con las del blanco muestras y establecer la concentración mínima a la cual el analito puede ser detectado y cuantificado de manera confiable. Generalmente se considera una relación señal / ruido entre o 10: 1 aceptable para estimar el límite de cuantificación.
3.-Basado en la desviación estándar de la respuesta y la pendiente: el límite de cuantificación (QL) puede expresarse como:
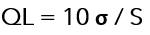
Donde:
σ = desviación estándar de la respuesta.
S = la pendiente de la curva de calibración.
La pendiente S puede estimarse a partir de la curva de calibración del analito.
-Basado en la curva de calibración: se debe estudiar una curva de calibración específica utilizando muestras que contengan un analito en el rango del Límite de Cuantificación. La desviación estándar residual de una línea de regresión o el estándar la desviación de las intersecciones con el eje y de las líneas de regresión pueden usarse como la desviación estándar.
Recomendaciones.
-El límite de cuantificación y el método utilizado para determinar el límite de cuantificación debe ser presentado, el límite debe validarse posteriormente mediante el análisis de un número adecuado de muestras conocidas o preparadas a la concentración del límite de cuantificación.
-La evaluación del parámetro de Limite de Cuantificación siempre se lleva a cabo con enfoque cuantitativo, es decir, tratar el ensayo como si fuese una valoración.
-El recobro no solo se puede ver afectado por la matriz también se ve alterado por la posible adhesión a componentes del contenedor / cierre, por ejemplo, vial de vidrio, válvula dosificadora. En general, un procedimiento de preparación de muestra más simple dará como resultado una menor variación de la recuperación.
No olvides que en caso de ser necesario, puedes contactarnos para mejorar el proceso de laboratorio y administrativo de tu organización a través software 100% confiable y seguro. Recuerda que en deappharma somos expertos en la materia, por lo que procesos complejos no representa un reto para nosotros, debido a que tenemos en mente los aspectos relevantes de cumplimiento.
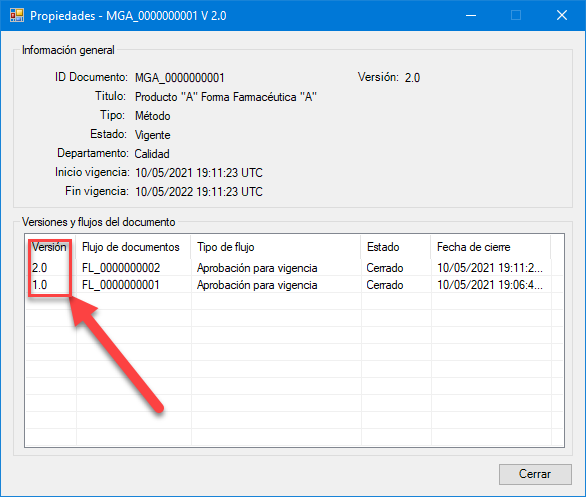
Proceso de linealidad del sistema y LOQ controlado y gestionado con eADMxL.
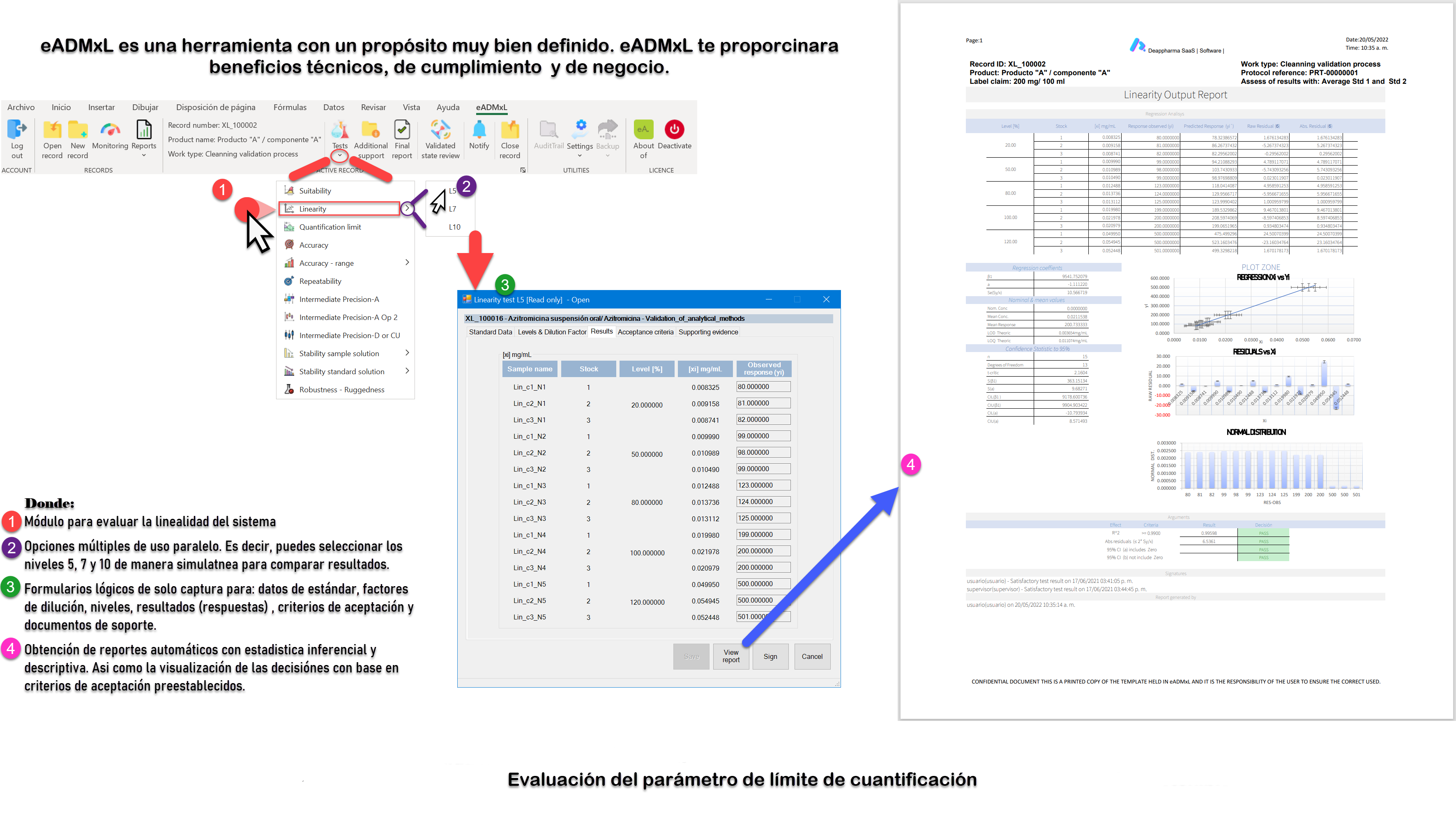
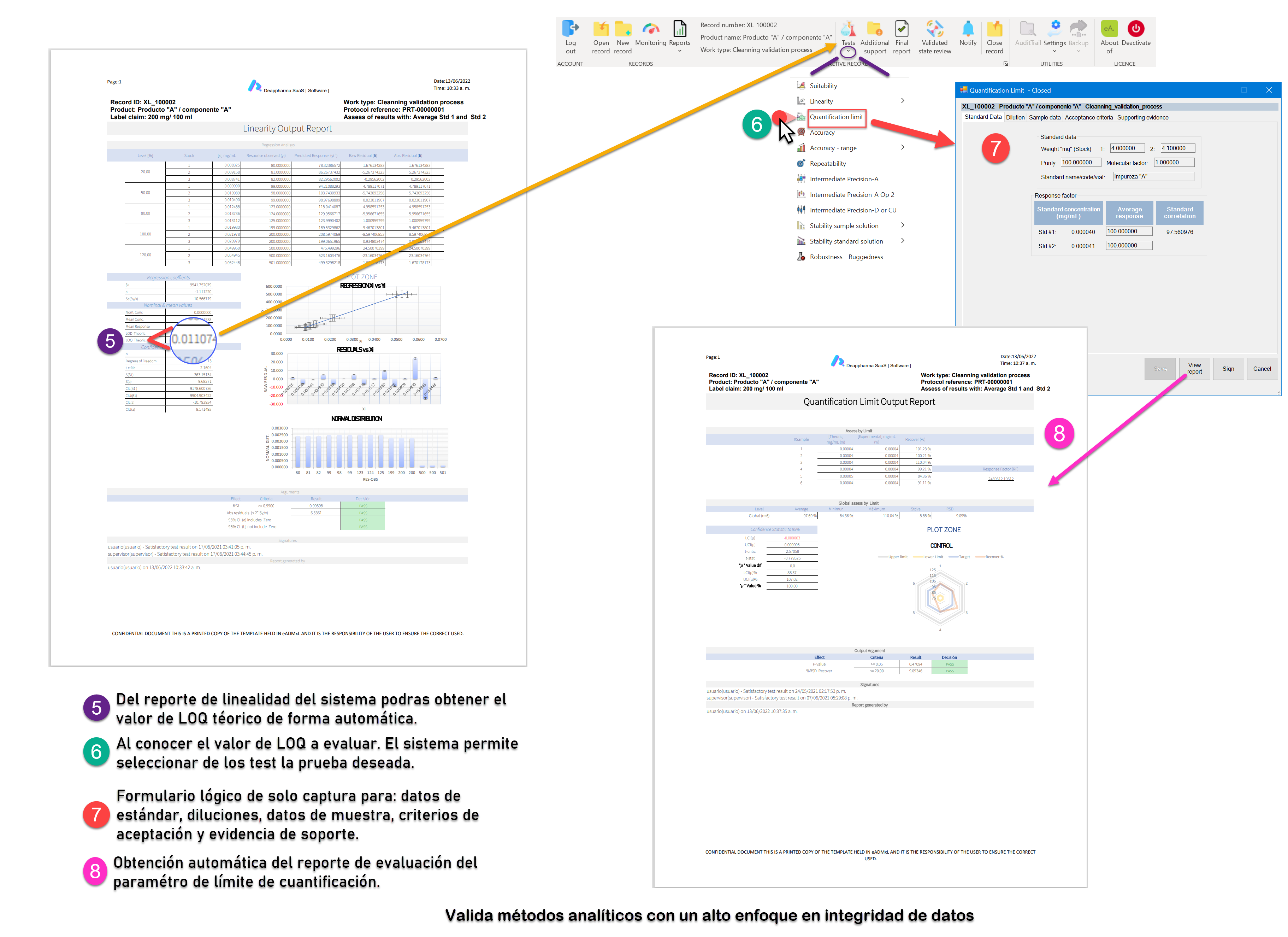
No olvides que, en caso de ser necesario, puedes contactarnos para mejorar el proceso de laboratorio y administrativo de tu organización a través software 100% confiable y seguro. Recuerda que en deappharma contamos con un software que te ayudara a controlar tu proceso de validación de métodos analíticos. Somos expertos en la materia, por lo que procesos complejos no representa un reto para nosotros, debido a que tenemos en mente los aspectos relevantes de cumplimiento.
Referencias.
- NORMA OFICIAL MEXICANA NOM-059-SSA1-2015, Buenas prácticas de fabricación de medicamentos.
- FDA analytical procedures and methods validation for drugs and biologics
- WHO guidelines on validation – appendix 4 analytical method validation
- ICH Harmonised tripartite guideline validation of analytical procedures: text and methodology q2(r1).
¡Contáctanos! y conoce de que manera te vamos a ayudar.
¡Únete a nuestra comunidad en redes!