11, Feb 2022
Audit Trail para asegurar y elevar el proceso de Integridad de datos
Este término regulatorio e informático se refiere a la auditoría, detección continua y exhaustiva de los datos. Por lo que es relevante contar con este proceso o función en cada uno de nuestros sistemas informatizados. El objetivo, es trazar la vida de un dato de manera perdurable.
La industria farmacéutica, dispositivos médicos y almacenes utilizan sistemas informatizados dentro de sus procesos. Por este hecho, es relevante el control y trazabilidad de datos. Para abordar el tema y conocer el alcance e importancia de la función de pistas de auditoria te invitamos a leer lo siguiente.
¿Qué es un “pista de auditoría”?
La «Pista de auditoría» o también conocida como «Audit Trail». Significa un registro electrónico seguro, generado por computadora con sellos de tiempo que permiten la reconstrucción del curso de los eventos relacionados con la creación, modificación y/o eliminación de un registro electrónico. Por ejemplo, la pista de auditoría para una ejecución de cálculos analíticos con la utilización de Excel como software de cálculo, debe incluir como mínimo lo siguiente: nombre de usuario, la fecha/hora de la ejecución, los cambios realizados sobre un valor y los detalles justificados de dicho cambio, según apliquen. ¡Pero no solo eso! Las pistas de auditoria son un aparte esencial de los sistemas computarizados. Es por esto, que los reportes de auditorita deben ser robustos y seguros.
Retomemos el ejemplo de uso de la hoja de Excel para obtención de resultados analíticos. En la actualidad la industria configura la obtención de pistas de auditoría sobre los mismos libros de Excel. Esta pista «LOG» regularmente la establecen dentro del mismo libro de manera anidada como una pestaña (hoja) de cálculo. ¡Esta práctica, es poco sostenible y segura! ya que la trazabilidad la dejas en manos de Excel y de la vulnerabilidad de código realizado con VBA.
¿Existe riesgo de perder la trazabilidad en mis libros de Excel por usar configuración VBA?
La respuesta es ¡Si! y tu impacto será alto porque no tendrás la manera de trazar la información.
Los riesgos los podemos clasificar en dos escenarios posibles:
- Daño en el archivo de Excel que no permita acceder a la información.
- Alteración y vulnerabilidad de la programación en VBA (Hasta videos en youtube te enseñan a desbloquear código relizadoo con VBA. ¡Ojo con esto!)
¿Quién debe revisar los registros de auditoría?
La revisión de la pista de auditoría es similar a la evaluación de las tachaduras en papel al revisar los datos. Personal responsable de la revisión de registros bajo cGMP debe revisar las pistas de auditoría que capturan los cambios en datos asociados con el registro a medida que revisan el resto del registro. Para ejemplo, todos los registros de producción y control, que incluyen registros de auditoría, deben revisarse y aprobado por la unidad de calidad. Las regulaciones brindan flexibilidad para tener algunas actividades revisadas por una persona que supervisa o verifica directamente la información.
FDA recomienda un enfoque de sistema de calidad para implementar la supervisión y revisión de cGMP registros.
¿Con qué frecuencia se deben revisar los registros de auditoría?
Si la frecuencia de revisión de los datos se especifica en las regulaciones de cGMP, adhiérase a esa frecuencia para la revisión de la pista de auditoría. Por ejemplo, requiere revisión después de cada paso significativo en fabricación, procesamiento, empaque o almacenamiento, y requiere la revisión de datos antes del lote liberar. En estos casos, aplicaría la misma frecuencia de revisión para la pista de auditoría.
Si la frecuencia de revisión de los datos no está especificada en las regulaciones de cGMP, debe determinar la frecuencia de revisión de la pista de auditoría utilizando el conocimiento de sus procesos y la evaluación de riesgos.
La evaluación de riesgos debe incluir la evaluación de la criticidad de los datos, los mecanismos de control e impacto en la calidad del producto. Los riesgos para los datos incluyen, entre otros, la posibilidad de que se eliminen modifique o excluyan sin autorización o sin detección.
Su enfoque para la revisión de la pista de auditoría y la frecuencia con la que la realiza deben garantizar que se cumplen los requisitos de cGMP, se implementan los controles apropiados y la confiabilidad de la revisión.
¿Puedo aumentar la trazabilidad de mis libros de Excel sin necesidad de usar VBA?
¡Si! y todo de manera virtual con el suso de nuestro complemento COM. Esto garantiza que tus datos sean trazados de manera perdurable.
En deappharma hemos desarrollado eDocuSeed una herramienta validada y especializada para el mantenimiento, gestión y trazabilidad de libros de Excel. ¡Sin duda te ayudaremos a mitigar tu riesgo! Puedes contactarnos para mejorar el proceso administrativo de tu organización a través software 100% confiable y seguro.
Referencias
- Data Integrity and Compliance With Drug CGMP Questions and Answers Guidance for Industry
- Guía para el enfoque de sistemas de calidad de la industria para las regulaciones CGMP farmacéuticas.
¡Contáctanos! y conoce de que manera te vamos a ayudar.
¡Únete a nuestra comunidad en redes!
- 0
- Por Team deappharma

24, Ene 2022
Detalles del Enfoque – Alcance de la Parte 11 de CFR
Detalles del Enfoque – Alcance de la Parte 11
Interpretación estrecha del alcance
Entendemos que existe cierta confusión sobre el alcance de la parte 11. Algunos han entendido que el alcance de la parte 11 es muy amplio. Creemos que algunas de esas amplias interpretaciones podrían conducir a controles y costos innecesarios y podrían desalentar la innovación y los avances tecnológicos sin brindar un beneficio adicional a la salud pública. Como resultado, queremos aclarar que la Agencia tiene la intención de interpretar el alcance de la parte 11 de manera restringida.
Según la interpretación restringida del alcance de la parte 11, con respecto a los registros que deben mantenerse según las reglas predicadas o presentarse a la FDA, cuando las personas optan por utilizar registros en formato electrónico en lugar de papel, se aplicaría la parte 11. Por otro lado, cuando las personas usan computadoras para generar impresiones en papel de registros electrónicos, y esos registros en papel cumplen con todos los requisitos de las reglas predicadas aplicables y las personas confían en los registros en papel para realizar sus actividades reguladas, la FDA generalmente no condicionara a las personas para «usar registros electrónicos en lugar de registros en papel». En estos casos, el uso de sistemas informáticos en la generación de registros en papel no activaría la parte 11.

Definición de registros de la Parte 11
Bajo esta interpretación estrecha, la FDA considera que la parte 11 es aplicable a los siguientes registros o firmas en formato electrónico (registros o firmas de la parte 11):
- Registros que deben mantenerse según los requisitos de la regla establecida y que se mantienen en formato electrónico en lugar del formato en papel. Por otro lado, los registros (y cualquier firma asociada) que no se requiera conservar según las reglas establecida, pero que, sin embargo, se mantienen en formato electrónico, no son registros de la parte 11.
Le recomendamos que determine, en función de las reglas establecidas, si los registros específicos son registros de la parte 11. Le recomendamos que documente tales decisiones.
- Registros que deben mantenerse según las reglas establecidas, que se mantienen en formato electrónico además del formato en papel, y en los que se confía para realizar actividades reguladas.
En algunos casos, las prácticas comerciales reales pueden dictar si está utilizando registros electrónicos en lugar de registros en papel. Por ejemplo, si se requiere que se mantenga un registro bajo una regla predicada y usted usa una computadora para generar una copia impresa en papel de los registros electrónicos, pero aun así depende del registro electrónico para realizar actividades reguladas, la Agencia puede considerar que usted está utilizando el registro electrónico en lugar del registro en papel. Es decir, la Agencia puede tener en cuenta sus prácticas comerciales para determinar si se aplica la parte 11.
En consecuencia, recomendamos que, para cada registro que deba mantenerse según las reglas establecidas, determine de antemano si planea confiar en el registro electrónico o en el registro en papel para realizar actividades reguladas. Recomendamos que documente esta decisión (por ejemplo, en un Procedimiento operativo estándar (SOP) o documento de especificación).
Registros enviados a la FDA, conforme a reglas establecidas (incluso si dichos registros no están específicamente identificados en las reglamentaciones de la Agencia) en formato electrónico. Sin embargo, un registro que no se presenta en sí mismo, pero que se utiliza para generar una presentación, no es un registro de la Parte 11 a menos que se requiera que se mantenga bajo una regla establecida y se mantenga en formato electrónico.
Firmas electrónicas que pretenden ser el equivalente de firmas manuscritas, iniciales y otras firmas generales requeridas por reglas establecidas. Las firmas de la Parte 11 incluyen firmas electrónicas que se utilizan, por ejemplo, para documentar el hecho de que ciertos eventos o acciones ocurrieron de acuerdo con la regla establecida (por ejemplo, aprobado, revisado y verificado).
Enfoque de los requisitos específicos de la Parte 11
Validación
La Agencia tiene la intención de ejercer la discreción de cumplimiento con respecto a los requisitos específicos de la parte 11 para la validación de sistemas computarizados y los requisitos correspondientes. Aunque las personas aún deben cumplir con todos los requisitos de las reglas establecidas aplicables para la validación, esta guía no debe interpretarse como una imposición de requisitos adicionales para la validación.
Sugerimos que su decisión de validar los sistemas computarizados y el alcance de la validación tengan en cuenta el impacto que los sistemas tienen en su capacidad para cumplir con los requisitos de las reglas establecidas. También debe considerar el impacto que esos sistemas podrían tener en la precisión, confiabilidad, integridad, disponibilidad y autenticidad de los registros y firmas requeridos. Incluso si no existe un requisito de regla de establecido para validar un sistema, en algunos casos puede ser importante validar el sistema.
Recomendamos que base su enfoque en una evaluación de riesgos justificada y documentada y una determinación del potencial del sistema para afectar la calidad y seguridad del producto y la integridad del registro. Por ejemplo, la validación no sería importante para un procesador de texto que se usa solo para generar SOP.
Pista de auditoría
La Agencia tiene la intención de ejercer la discreción de cumplimiento con respecto a los requisitos específicos de la parte 11 relacionados con las pistas de auditoría generadas por computadora y con sello de tiempo y cualquier requisito correspondiente. Las personas aún deben cumplir con todos los requisitos de reglas establecidas aplicables relacionados con la documentación de, por ejemplo, fecha, hora o secuencia de eventos, así como cualquier requisito para garantizar que los cambios en los registros no oscurezcan entradas previas.
Incluso si no existen requisitos de reglas establecidas para documentar, por ejemplo, la fecha, la hora o la secuencia de eventos en una instancia particular, puede ser importante contar con pistas de auditoría u otras medidas de seguridad físicas, lógicas o de procedimiento para garantizar la confiabilidad y confiabilidad de los registros. Le recomendamos que base su decisión de aplicar pistas de auditoría u otras medidas apropiadas, en la necesidad de cumplir con los requisitos de la regla establecida, una evaluación de riesgos justificada y documentada, y una determinación del efecto potencial sobre la calidad y seguridad del producto y la integridad del registro. Le sugerimos que aplique los controles apropiados basados en dicha evaluación. Los registros de auditoría pueden ser particularmente apropiados cuando se espera que los usuarios creen, modifiquen o eliminen registros regulados durante el funcionamiento normal.
Sistemas heredados
La Agencia tiene la intención de ejercer la discreción de ejecución con respecto a todos los requisitos de la parte 11 para los sistemas que de otro modo estaban operativos antes del 20 de agosto de 1997, la fecha de vigencia de la parte 11, bajo las circunstancias especificadas a continuación.
Esto significa que la Agencia no tiene la intención de tomar medidas de cumplimiento para imponer el cumplimiento de los requisitos de la parte 11 si se cumplen todos los siguientes criterios para un sistema específico:
- El sistema estaba operativo antes de la fecha de vigencia.
- El sistema cumplió con todos los requisitos de reglas establecidas aplicables antes de la fecha de vigencia.
- Actualmente, el sistema cumple con todos los requisitos de reglas establecidas aplicables.
- Tiene evidencia documentada y justificación de que el sistema es apto para el uso previsto (lo que incluye tener un nivel aceptable de seguridad e integridad de registros, si corresponde).
Si un sistema ha cambiado desde el 20 de agosto de 1997, y si los cambios impiden que el sistema cumpla con los requisitos de la regla establecidad, los controles de la Parte 11 deben aplicarse a los registros y firmas de la Parte 11 de conformidad con la política de cumplimiento expresada en esta guía.
Copias de Registros
La Agencia tiene la intención de ejercer la discreción de ejecución con respecto a los requisitos específicos de la parte 11 para generar copias de registros y cualquier requisito correspondiente. Debe proporcionar a un investigador un acceso razonable y útil a los registros durante una inspección. Todos los registros en su poder están sujetos a inspección de acuerdo con las reglas establecidas.
Recomendamos que proporcione copias de registros electrónicos por:
- Producir copias de registros mantenidos en formatos portátiles comunes cuando los registros se mantienen en estos formatos
- Usar métodos de conversión o exportación automatizados establecidos, cuando estén disponibles, para hacer copias en un formato más común (los ejemplos de dichos formatos incluyen, entre otros, PDF, XML o SGML)
En cada caso, recomendamos que el proceso de copiado produzca copias que conserven el contenido y el significado del registro. Si tiene la capacidad de buscar, ordenar o buscar tendencias en los registros de la parte 11, las copias entregadas a la Agencia deben proporcionar la misma capacidad si es razonable y técnicamente factible. Debe permitir la inspección, revisión y copia de registros en un formato legible en su sitio utilizando su hardware y siguiendo sus procedimientos y técnicas establecidos para acceder a los registros.
Retención de registros
La Agencia tiene la intención de ejercer la discreción de ejecución con respecto a los requisitos de la parte 11 para la protección de registros para permitir su recuperación precisa y rápida durante todo el período de retención de registros y cualquier requisito correspondiente. Las personas aún deben cumplir con todos los requisitos de reglas establecidas aplicables para la retención y disponibilidad de registros.
Sugerimos que su decisión sobre cómo mantener los registros se base en los requisitos de las reglas predicadas y que base su decisión en una evaluación de riesgos justificada y documentada y en una determinación del valor de los registros a lo largo del tiempo.
FDA no tiene la intención de objetar si usted decide archivar los registros requeridos en formato electrónico en medios no electrónicos como microfilm, microficha y papel, o en un formato de archivo electrónico estándar (ejemplos de tales formatos incluyen, entre otros, PDF, XML o SGML). Las personas aún deben cumplir con todos los requisitos de las reglas establecidas, y los propios registros y cualquier copia de los registros requeridos deben conservar su contenido y significado. Siempre que los requisitos de la regla establecida se cumplan por completo y el contenido y el significado de los registros se conserven y archiven, puede eliminar la versión electrónica de los registros. Además, los componentes de firmas y registros en papel y electrónicos pueden coexistir (es decir, una situación híbrida) siempre que se cumplan los requisitos de la regla establecida y se conserve el contenido y el significado de esos registros.
No olvides que en caso de ser necesario, puedes contactarnos para mejorar el proceso de laboratorio y administrativo de tu organización a través software 100% confiable y seguro. Recuerda que en deappharma somos expertos en la materia, por lo que procesos complejos no representa un reto para nosotros, debido a que tenemos en mente los aspectos relevantes de cumplimiento.
REFERENCES
- Glossary of Computerized System and Software Development Terminology (Division of Field Investigations, Office of Regional Operations, Office of Regulatory Affairs, FDA 1995)
- General Principles of Software Validation; Final Guidance for Industry and FDA Staff (FDA, Center for Devices and Radiological Health, Center for Biologics Evaluation and Research, 2002)
- Guidance for Industry, FDA Reviewers, and Compliance on Off-The-Shelf Software Use in Medical Devices (FDA, Center for Devices and Radiological Health, 1999)
- Pharmaceutical CGMPs for the 21st Century: A Risk-Based Approach; A Science and Risk-Based Approach to Product Quality Regulation Incorporating an Integrated Quality Systems ApproachExternal Link Disclaimer (FDA 2002)
¡Contáctanos! y conoce de que manera te vamos a ayudar.
¡Únete a nuestra comunidad en redes!
12, Ene 2022
Ciclo de vida de la información
En la actualidad las instituciones y empresas producen, almacenan y respaldan gran cantidad e información. Hoy en día, la información que posee una empresa es uno de sus valores mas importantes. Este valor no solo es mantener un acceso rápido a los dispositivos que la almacenan. Existen casos donde no se justifica el gasto porque el valor de la información no siempre es el mismo. El veloz crecimiento del volumen de información no puede ser excusa para no organizar esta información según su criticidad y volumen. Cuando la información de una empresa o institución no se encuentra ordenada y clasificada se producen inconvenientes que dificultan su procesamiento ya que se presentan diferentes tiempos de acceso a la información.
El concepto de ciclo de vida esta evolucionado rápidamente en la industria de la información (medianas y grandes empresas) junto al valor que le otorga la información. La idea que sustenta el ciclo de vida es el concepto de la cadena de valor que posee una información en particular desde que se crea hasta que se accede a ella en mayor o menor medida. En estos accesos pasa por una serie de procesos cuya función es añadir valor para quien debe utilizarla hasta agotar su funcionalidad. Sucede que al aumentar el valor de la información también se aumenta el coste de almacenamiento.
Los inconvenientes mencionados surgen ya que el desorden o falta de clasificación se produce por no acceder a la información por los mismos canales o bajo las mismas pautas y los detallamos a continuación.
1.1-Dificultad de acceder a llegar a la información
Mencionábamos que una organización depende de la información que tiene almacenada para poder llevar a cabo sus objetivos. Que el volumen de información esté informatizado no implica que esté de forma ordenada, es decir, que los integrantes de la información sepan cómo, o tengan los medios necesarios para acceder a la información. Éste es el primer obstáculo a superar cuando hablamos de gestionar información dentro del ciclo de vida.
1.2.-Deficiencia en el acceso en cuanto a tiempo y forma
Superado el primer inconveniente, nuestra tarea continua en mejorar el acceso a la información considerando los aspectos técnicos. En esta tarea lo principal es el análisis de los requerimientos de la organización y del relevamiento del hardware disponible para llevar adelante las operaciones.
1.3.-Diferentes modos de acceder a la información
Por último, pondremos foco en mejorar el acceso a la información teniendo presente los aspectos externos al hardware y dependientes de cada uno de los usuarios o áreas que necesitan acceder.
Muchas veces para mitigar o corregir parcialmente estos inconvenientes se intenta alojar la mayor cantidad de información en servidores con discos duros (de alto rendimiento). Ya que el alojamiento en estos servidores ayudan a mantener, gestionar, hacer peticiones y establecer conexiones de bases de datos que ayudan con el orden de información.
Ya hemos dicho que el significado y la importancia de esta información serán los mismos a medida que transcurre el tiempo. Debido a esto la forma, los métodos y las técnicas con que debe ser almacenada y respaldada la información cambiaran de acuerdo al paso del tiempo, ya que la información no tendrá el mismo valor, es decir, el concepto principal que obtenemos del ciclo de vida de la información es almacenar la información donde podremos obtener el mejor beneficio de ella.
Relevar y analizar los puntos antes mencionados nos proporcionaran una idea muy acertada de cómo encarar al almacenamiento y, si fuera necesario, protección (backups) de la información concerniente al sistema que estamos analizando, diseñado o administrando.
No olvides que en caso de ser necesario, puedes contactarnos para mejorar el proceso de laboratorio y administrativo de tu organización a través software 100% confiable y seguro. Recuerda que en deappharma somos expertos en la materia, por lo que procesos complejos no representa un reto para nosotros, debido a que tenemos en mente los aspectos relevantes de cumplimiento.
Referencia;
- Administración de Storage y Backups_Dante cantone, España, Editorial Alfa Omega
¡Contáctanos! y conoce de que manera te vamos a ayudar.
¡Únete a nuestra comunidad en redes!
20, Dic 2021
Hojas de cálculo con integridad de datos con cumplimiento CFR21
Las industrias reguladas utilizan las hojas de cálculo para muchos propósitos. Haciendo que Excel cubra diferentes necesidades atribuidas a las actividades de rutina. Este programa «Excel» puede: calcular, organizar y analizar datos, de tal manera que pueda evaluar el funcionamiento de: procesos, métodos, tendencias y en algunas ocasiones la obtención de documentos, con base en plantillas que se alimentan de bases de datos creadas propiamente en la misma aplicación. Las hojas de cálculo se configuran en un formato de cuadrícula. Cada pieza de información se coloca en su propia celda para que pueda ser usada adecuadamente. Los datos pueden ser números o palabras. Las palabras se organizan alfabéticamente y los números numéricamente. Los números se calculan con suma, resta, multiplicación, división y en algunos casos estadística más avanzada. Partiendo de esto , el uso de hojas de cálculo dentro de la industria regulada toma relevancia, ya que impacta en mayor medida sobre procesos de carácter GxP.
La situación actual
La versatilidad de las hojas de cálculo proporciona opciones de diferentes alcances GxP, por ejemplo;
Bases de datos, con el siguiente alcance:
- Control de muestras
- Control de resultados de resultados de laboratorio
- Listado de muestreos de materiales
- Listado de muestra de retención
- Listado de estatus de documento
- Asignación de código de documentos
- Listados maestros de estabilidades
- Listados de toma de muestras para programa anual, entre otros.
Hojas de cálculo, con el siguiente alcance:
- Emisión de resultados de: ensayo, uniformidad de contenido, sustancias relacionadas, disolución
- Evaluación de todos parámetros de desempeño del proceso de validación de métodos analíticos
- Evaluación de resultados de perfiles de disolución, entre otros.
Plantillas, con el siguiente alcance:
- Emisión de certificados de análisis
- Emisión de protocolos
- Emisión de reportes, entre otros.
Derivado del dinamismo y la versatilidad de la aplicación Excel, podemos obtener diferentes magnitudes de hallazgos relacionadas a eventos de integridad de datos, tales cuales ponen en cuestionamiento el cumplimiento regulatorio.
Las situaciones relevantes por el uso de hojas de cálculo son los siguientes:
Desarrollo
- Desarrollo de plantillas con esquemas de reedición
- Desarrollo de plantillas en los que no ha sido retada la estadística contenida en estas
- Desarrollo de fórmulas estadísticas establecidas, sin una base de conocimiento estadístico.
Funcionalidad
- No cuentan con perfiles robustos para el ingreso a la plantilla, niveles de usuario y definición del alcance de estos para el desempeño en el uso de mismas
- No hay validación de las celdas, discriminando si son numéricas o textuales
- Seguridad de las hojas solo estableciendo contraseñas con las propiedades de Excel o con métodos poco robustos como VBA (Visual Basic Application)
- No se puede justificar y atribuir a alguien el cambio y/o modificación de un dato-valor
- La seguridad de la hoja se da con el bloqueo de la misma
- Se pueden copiar y pegar las hojas en otras rutas
- Uso de hojas multiusuario.
Integridad de datos
- No cuentan con Audit trail “pistas de auditoria” (elemento crítico para dar trazabilidad a cada movimiento realizado en un entorno GxP computarizado)
- No cuentan con la capacidad de respaldo y recuperación de la información
- Los datos no se asocian con bases de datos que hagan que estos perduren y, por tanto no pueden ser mantenidos; Atribuibles,legibles, contemporáneos y exactos (ALCOA).
Principio
Derivado de los puntos y controles sugeridos por las agencias regulatorias con el fin de garantizar la integridad de datos y con esto mantener el concepto de “ALCOA ++” partiendo de que los datos sean perdurables, se necesitan infraestructuras para garantizar la misma.
ALCOA ++, se atribuye a los siguientes términos:
- Atribuible, todas las actividades tales como crear, borrar o modificar son relacionadas a un individuo.
- Legible, la información debe ser clara y visible.
- Contemporáneo, la información debe ser registrada en el momento de hacer la actividad.
- Original, los datos deben provenir de registros originales o copias fieles a la original.
- Exactos, la exactitud debe ser controlada para eliminar errores y los cambios deben ser documentados.
- +Completo, todos los datos pertenecientes al registro GxP deberán ser retenidos.
- +Consistente, la línea del tiempo del metadato debe ser lógico.
- +Duradero, todos los registros deben estar en un medio duradero de manera legible durante su tiempo de retención.
- +Disponible, los datos deberán estar disponibles y accesibles en su totalidad.
Qué refiere una hoja de cálculo con integridad?
Las hojas de cálculo con integridad de datos, manifiestan un alto comportamiento en la trazabilidad y vida de un dato-valor, de tal manera que la perdurabilidad de este sea consistente y por tanto se pueda mantener: atribuible, legible, contemporáneo y exacto.
Suponga que está utilizando una hoja de cálculo y que necesita cambiar los valores obtenidos de al menos una prueba dentro de una fila. Tiene un pequeño problema: tiene que cambiar el valor de varias filas. Por ejemplo, el pH aparece en tres filas. Pero aparecerá un problema de verdad si olvida cambiar uno de los valores de las filas – el valor de pH asignado a esta fila se volverá ambigua, y por tanto sus datos perderán integridad.
Como hacer que una hoja de cálculo tenga Integridad de datos?
Las hojas de cálculo se deben evaluar y, con base en esto determinar el alcance GxP.

Derivado de la determinación, podemos considerar el diseño del sistema computarizado. Para esta transformación se necesitan procesos de programación mediante lenguajes robustos obtenidos por aplicaciones. Las hojas de cálculo más recientes con enfoque a entornos y control de datos GxP tiene estas capacidades.
CFR21 parte 11.10 «Controles»
Cuando hacemos referencias a hojas de cálculo, estas deben ser tratadas como un sistema computarizado. Por este hecho a esta herramienta le aplican los siguientes controles regulatorios y de cumplimiento:
- Debe permitir el proceso de validación para garantizar la precisión, la confiabilidad, el desempeño esperado consistente y la capacidad de discernir registros inválidos o alterados.
- Debe proporcionar la capacidad de generar copias precisas y completas de los registros en forma legible y en formato electrónico, para que la entidad reguladora pueda inspeccionarlos, revisarlos y copiarlos.
- Debe permitir la recuperación precisa , durante todo el período de retención de registros.
- Debe limitar el acceso del sistema a personas autorizadas.
- Debe usar pistas de auditoría seguras, generadas por computadora y con marca de tiempo, para registrar de manera independiente la fecha y la hora de las entradas y acciones del operador que crean, modifican o eliminan registros electrónicos. Los cambios de registro no deben ocultar la información previamente grabada.
- Debe permitir establecer el cumplimiento de políticas escritas, que responsabilizan a las personas de las acciones iniciadas bajo sus firmas electrónicas, con el fin de impedir la falsificación de registros y firmas.
- Debe proporcionar controles de autoridad, para garantizar que solo las personas autorizadas puedan usar el sistema, firmar electrónicamente un registro, acceder a la operación o al dispositivo de entrada o salida del sistema informático, alterar un registro o realizar operación en cuestión.
- Debe tener controles adecuados sobre la distribución, el acceso y el uso de la documentación para la garantizar su trazabilidad.
Por qué es importante la integridad de datos?
La importancia radica en disminuir el riesgo de que los datos críticos pueden ser falseados, borrados, divulgados sin autorización, modificados o negados por los emisores. Y con esto, no tener la posibilidad de trazar la historia y/o auditarla.
Lineamientos regulatorios a considerar
Todos los registros de calidad y técnico/científicos (incluyendo informes de análisis, certificados de análisis y hojas de trabajo analítico) deben ser legibles, rápidamente recuperables, almacenados y retenidos dentro de instalaciones que proporcionen un medio ambiente adecuado que prevenga modificaciones, daño o deterioro y/o pérdida.
Las condiciones bajo las cuales todos los registros originales son almacenados deben ser tales que aseguren su seguridad y confidencialidad, y el acceso a ellos debe estar restringido al personal autorizado. Se pueden emplear también almacenamiento y firmas electrónicos, pero con acceso restringido y en conformidad con los requisitos de los registros electrónicos.
Disposiciones asociadas a sistemas cerrados (incluye hojas de Excel)
- Limitar el acceso al sistema a personas autorizadas.
- Uso de verificaciones del sistema operativo.
- Uso de controles de autoridad.
- Determinación de que las persona que desarrollan , mantienen o usas sistemas tienen la educación , capacitación y experiencia para realizar las tareas asignadas.
- Establecimiento de políticas escritas que responsabilizan a las personas por las acciones iniciadas bajo sus firmas electrónicas.
- Controles apropiados sobre la documentación de los sistemas.
No olvides que en caso de ser necesario, puedes contactarnos para mejorar el proceso de laboratorio y administrativo de tu organización a través software 100% confiable y seguro. Recuerda que en deappharma somos expertos en la materia, por lo que procesos complejos no representa un reto para nosotros, debido a que tenemos en mente los aspectos relevantes de cumplimiento. ¡Conoce nuestras soluciones da clic aquí!
Referencias
- CFR 21 Parte 11.10 Closed systems
- FDA Data Integrity and Compliance With CGMP Guidance for Industry
- NOM 059 2015 Buenas prácticas de fabricación de medicamentos.
- WHO Technical Report Series, No. 957, 2010 44th Report – Annex 1 Buenas prácticas de la OMS para laboratorios de control de calidad de productos farmacéuticos
¡Contáctanos! y conoce de que manera te vamos a ayudar.
¡Únete a nuestra comunidad en redes!
2, Dic 2021
Buenas prácticas de documentación digital.
A partir del desarrollo de las tecnologías de la información se han descubierto campos inéditos como el de la documentación en soporte digital que, sin duda, a planteado nuevos retos en torno a su administración, acceso y preservación. De hecho, a causa del aumento de documentos en soporte digital, tanto la archivística como la tecnología han visto la necesidad de desarrollar políticas, estrategias y actividades tendientes a enfrentar este nuevo desafío, el cual, reclama acciones inmediatas y en la medida de lo posible soluciones estandarizadas a nivel internacional.
Qué es un documento digital?
Desde una perspectiva terminológica, sostiene que, el documento digital son datos o información que han sido capturados y fijados para su almacenamiento y manipulación en un sistema automatizado y que requieren del uso de este para hacerlo accesible a una persona. Se puede decir entonces que, este tipo de documentos son producidos a través de procesadores de texto, hojas de cálculo y gráficos que constituyen los resultados de distintos programas o paquetes de software. De allí la importancia de estos programas para la visualización de los documentos digitales debido a que actúan como un intermediario entre el hardware y la persona que está accediendo a la información
Una perspectiva diferente la proporciona el Modelo de Requisitos para la Gestión de Documentos Electrónicos de Archivo , que lo define como un documento que está en forma electrónica/digital porque ha sido creado mediante un programa informático o bien porque se ha digitalizado por ejemplo, cuando se ha escaneado a partir de un documento en papel. Asimismo, se puede puntualizar como un documento analógico o digital que es transportado por un conducto eléctrico y requiere del uso de equipamiento para ser inteligible por parte de una persona. En consecuencia y comparando las anteriores concepciones sobre el documento digital, coinciden en que se trata de información que se encuentra en soporte digital, que además posee un conjunto de códigos que precisan de una herramienta de tipo eléctrico para ser decodificado con el fin de hacerlo accesible al usuario. Son entonces, documentos que han sido creados en un entorno digital y que para su reproducción y accesibilidad requieren de un hardware (partes tangibles de un sistema informático) y de igual modo, un software (componentes lógicos de un sistema informático) Sin embargo, no basta una perspectiva netamente instrumental haciendo referencia a los elementos físicos que facilitan la visualización de los documentos digitales, existe además, un componente lógico señalando que este tipo de documentos “contienen un mensaje de texto alfanumérico, conformado por los bits, formado por ceros y unos que se procesan sobre un soporte, llámese cinta o disco (en general, soporte electrónico) que está en principio destinado a durar en el tiempo.”. Con base en lo anterior, se puede afirmar que este tipo de documentos poseen características que los diferencian de aquellos producidos en soporte papel, esencialmente, por el uso de símbolos que se registran en un soporte y en un medio al que las personas no pueden acceder de manera directa por lo cual es necesario descifrarlos.
A partir de estas perspectivas sobre el documento digital, parece poder deducirse de forma argumentativa que lo que distingue a este documento digital de un documento en soporte papel es que en su gestión interviene de forma directa la tecnología. El documento digital si bien es diferente en el soporte, comparte con el documento en papel una misma función.
Preservación digital
Como consecuencia de la producción masiva de documentos digitales, surge simultáneamente la necesidad crear estrategias que permitan asegurar su preservación a largo del tiempo. En otras palabras, las estrategias de preservación documental ya no se relacionan únicamente con el soporte papel y otros documentos tangibles sino que también debe contemplar el tratamiento de los documentos digitales. En este sentido “la preservación digital comprende acciones específicas cuyo fin ulterior y a largo plazo es asegurar la permanencia y acceso del contenido de documentos digitales a lo largo del tiempo y las tecnologías, independientemente de su soporte, formato o sistema”. Para llevar a cabo este propósito es necesario emprender actividades que contemplen tanto su mantenimiento y protección, como su resguardo de manera anticipada para prevenir futuros deterioros o daños en la información. Se busca principalmente que existan los medios para identificar, mantener y preservar para siempre la información registrada pues constituyen el registro del pasado, permite entender el mundo actual, y sienta las bases para la construcción permanente de conocimiento.
El contenido informativo de los documentos digitales es uno de los aspectos sobre los cuales están dirigidos los esfuerzos de la preservación digital, ya que la preservación supone llevar a cabo tareas como la conservación preocupándose no solo por el mantenimiento del objeto, sino también, y principalmente por su contenido informativo.
Por esta razón la preservación de los documentos digitales centra su desarrollo en mantener la capacidad de visualizar, recuperar y utilizar de forma adecuada la información, sin dejar de lado, las frecuentes transformaciones que sufre el entorno tecnológico. Incluso en un sentido tradicional, la preservación ha estado enfocada a la forma física de los documentos, sin embargo, aspectos como el desarrollo de la tecnología, la aparición del documento digital, el alcance y concepto que se conoce como preservación documental ha necesitado una evolución y adaptación que va más allá de la integridad física del objeto para intervenir en la integridad intelectual y el aseguramiento de la información con todos sus elementos asociados.
Se habla del mantenimiento a largo plazo de la cadena de bits y la accesibilidad continuada del contenido. De manera que es importante también determinar estrategias que faciliten no solo la preservación del soporte, es decir el hardware que permite la visualización de la información, sino que garanticen de igual modo, el contenido del documento, esto es, la información contenida, que puede trascender a las generaciones para ser consultada por cualquier persona independientemente del dispositivo que utilice para ello. La preservación entonces, busca soluciones que permitan la permanencia y accesibilidad a la infinita cantidad de información digital que se está produciendo hoy en día.
La preocupación surge al cuestionarse sobre si esta información digital llegará a las generaciones venideras, siendo más profundo el problema al evaluar la fragilidad de los soportes y el frecuente cambio en las tecnologías. Esta perspectiva es señalada donde la preservación digital se define como las actividades necesarias para asegurar el acceso continuado a materiales digitales hasta cuando sea necesario, a pesar de los obstáculos que representan los fallos en los soportes o los cambios tecnológicos. Los planteamientos anteriormente señalados coinciden en dos aspectos fundamentales que debe observar la preservación digital. El primero sobre aquellas acciones, procedimientos, directrices o procesos que posibiliten de forma ilimitada el acceso a la información independiente del soporte y el contenido. En otras palabras, la preservación digital no existiría si no se mantiene la posibilidad de acceder a los recursos digitales. El segundo aspecto hace referencia a la disponibilidad de estos documentos a lo largo del tiempo. Se habla entonces de preservación a largo plazo la cual garantiza que la información pueda ser consultada en los próximos años superando obstáculos como la obsolescencia de los soportes y la evolución de la tecnología. Finalmente, estas labores de preservación deben implementarse teniendo en cuenta elementos cambiantes como el deterioro de los medios físicos y tecnológicos a través de los cuales se gestionan los contenidos digitales.
Como conclusión se puede decir que la preservación digital hace referencia al conjunto de estrategias tendientes a conservar la información que se encuentra en formato digital bien sean estos documentos, fotografías, archivos de audio, bases de datos entre otros. Su objetivo es facilitar y perfeccionar acciones tan importantes como el: acceder, visualizar, descifrar, interpretar, comprender e interactuar con documentos u objetos digitales de forma valida y sencilla con el propósito de facilitar la transferencia de datos e información que aseguren la gestión de conocimiento en concordancia con la evolución de la humanidad. La preservación a largo plazo aplica a documentos electrónicos y digitales independientemente del formato, y requiere a su vez, de la elaboración de un plan de preservación a largo plazo que incluyen acciones para asegurar además su conservación, su acceso y disponibilidad a lo largo del tiempo. También es importante mencionar que el almacenamiento digital es fácil, sin embargo, la preservación no lo es. Preservar digitalmente la información almacenada, organizada, conlleva no solo una planeación estratégica como política institucional, requiere a su vez una serie de gastos emanados de las transformaciones y/o actualizaciones tecnológicas tanto del software como de los hardware presentes en la sociedad del conocimiento que evoluciona con rapidez.
Buenas prácticas para la preservación digital
En la sociedad actual las organizaciones están creando, recibiendo y gestionando continuamente documentos digitales, lo cual, ha suscitado nuevos retos para las administraciones teniendo en cuenta el alto valor que representan la información en medio digital no solo para la toma de decisiones, sino para la garantizar la conformación y preservación del patrimonio digital institucional. Se habla entonces de las buenas prácticas para la gestión de documentos digitales los cuales poseen características especiales que exigen acciones específicas para garantizar su preservación durante el tiempo que sea necesario. Desde una perspectiva legal el Archivo General de La Nación (2014), en su compilación normativa establece que “una norma de buenas prácticas es un documento que nos indica cómo actuar siguiendo metódicamente unas pautas establecidas, reconocidas y aceptadas por un colectivo profesional o social suficientemente representativo y aprobado.” (p. 8). En este concepto se identifican elementos que reafirman la complejidad de la preservación digital pues si se habla de buenas prácticas, previamente a la conformación y divulgación de las mismas, es necesario que un grupo interdisciplinar de profesionales al interior de las organizaciones establezcan y aprueben de forma conjunta los procedimientos que guiaran las labores de preservación digital institucional. En efecto, la preservación digital surge como respuesta a un problema de carácter tecnológico que no debe ser abordado desde esta perspectiva únicamente. Dentro de una organización la preservación digital incluye aspectos administrativos, financieros y legales los cuales, en conjunto, le dan forma a un programa de preservación que facilitará la consolidación del patrimonio documental institucional. Las buenas prácticas de preservación digital deben estar enmarcadas en una política institucional que cuente con un firme respaldo financiero, un apoyo a nivel gerencial y por supuesto, un compromiso administrativo por parte de los funcionarios que intervienen en la cadena de preservación. Por este motivo, la definición de las buenas prácticas debe contemplar la resolución de preguntas como ¿Que guardar y porqué guardarlo?, ¿Dónde guardarlo?, ¿Hasta cuándo guardarlo?, ¿Cómo encontrarlo después?, ¿Cómo hacer que se mantenga inalterado?, ¿Cómo evitar que se vuelva obsoleto?
No olvides que en caso de ser necesario, puedes contactarnos para mejorar el proceso de laboratorio y administrativo de tu organización a través software 100% confiable y seguro. Recuerda que en deappharma somos expertos en la materia, por lo que procesos complejos no representa un reto para nosotros, debido a que tenemos en mente los aspectos relevantes de cumplimiento.
¡Contáctanos! y conoce de que manera te vamos a ayudar.
¡Únete a nuestra comunidad en redes!
29, Jun 2021
Revisión de estado validado de método analíticos.
Uno de los varios procesos de control analítico dentro de la industria, es el seguimiento o mantenimiento del estado validado y/o estado de control de métodos analíticos. No es una tarea fácil!! ya que para realizar una correcta revisión, el entorno en el que gira el proceso de mantenimiento validado radica en el acceso a documentos y en la obtención de datos que se asocian directamente a la esencia del método, como son: primera validación y parámetros de adecuabilidad históricos.
Concepto:
El concepto de ciclo de vida, vincula el desarrollo de productos y procesos, por lo que se debe adquirir conocimiento para abordar la revisión.
Validación métodos y calidad de los medicamentos.
La validación efectiva del método contribuye significativamente a asegurar la calidad de los medicamentos. Lo básico, es comprender el principio de garantía de calidad. Este principio describe que se debe producir un medicamento que sea apto para el uso previsto.
Este principio incorpora el entendimiento de que existen las siguientes condiciones:
a) La calidad, la seguridad y la eficacia están diseñadas o integradas en el producto.
b) La calidad no puede garantizarse adecuadamente simplemente mediante el producto en proceso y terminado, inspección o prueba.
Un programa de validación exitoso depende de la información y el conocimiento del método, así como, de la comprensión del proceso de desarrollo analítico. Este conocimiento es la base para establecer un enfoque para el control del analítico, que da como resultado certidumbre en la calidad y atributos evaluados. Los científicos analíticos deben:
- Comprender las fuentes de variación (se puede evaluar durante la validación o con la revisión de los datos de validación).
- Detectar la presencia y el grado de variación (análisis de datos obtenidos en la validación y en el histórico de datos de liberación, sin embargo, esto no mide directamente el método).
- Controlar la variación de una manera acorde con el riesgo, que representa para el método que afecta los datos de liberación del producto.
Descripción del proceso analítico en tres etapas:
Etapa 1 – Diseño del proceso analítico: el proceso analítico se define durante este etapa, basada en el conocimiento adquirido a través de actividades de desarrollo.
Etapa 2 – Validación del proceso analítico: durante esta etapa, el diseño analítico se evalúa para determinar si método es capaz de producir resultados reproducibles.
Etapa 3 – Verificación continua del proceso analítico: la garantía continua se obtiene durante la rutina analítica, de tal manera, que se pueda verificar que el proceso permanece en un estado de control.
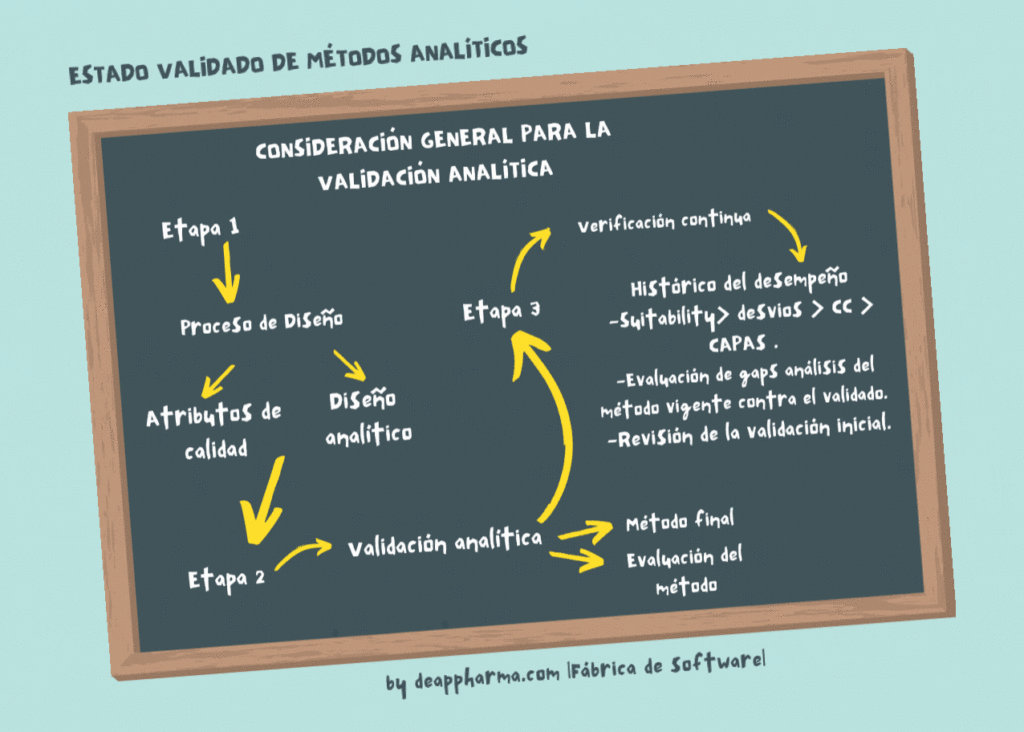
Los puntos relevantes de evaluación a la etapa 3, son:
- Requisitos de idoneidad históricos del sistema (platos teóricos, resolución, factor de coleo, factor de simetría, factor de capacidad y/o tiempos de retención de componentes).
- Parametrización y evaluación de las pruebas de desempeño acordes al método analítico.
- Revisión de los ajustes a las condiciones de operación indicadas en el procedimiento analítico vigente vs los descritos en la validación y/o transferencia inicial.
- Hallazgos o desviaciones, directamente relacionadas al desempeño del método analítico.
Si una evaluación basada en riesgos es utilizada como herramienta, se deben considerar otros factores. Los cuales pueden conducir a cambios en un procedimiento analítico y/o reemplazo con un nuevo método, se puede considerar los siguiente:
- Un ejercicio nuevo de transferencia;
- Una posible revalidación, sea parcial o total, o,un nuevo ejercicio de validación o,
- Un estudio de comparabilidad de métodos analíticos, es una opción factible.
En algunos casos, cambios en el fármaco o sustancias dentro del proceso de fabricación también deben ser evaluados como parte de la revisión de estado validado.
Recurso analítico «software».
Como se menciono anteriormente, la revisión de estado validado de un método analítico radica en que tan disponible y correcta fue la ejecución de este. Por lo que, la disposición de la información debe ser consistente. De otra manera será difícil concluir.
eADMxL el software de cumplimiento de deappharma.com , le ayudara con el control del proceso de estado validado mediante sus funciones especificas de «Revisión de estado validado» y «Monitoreo de vigencias».
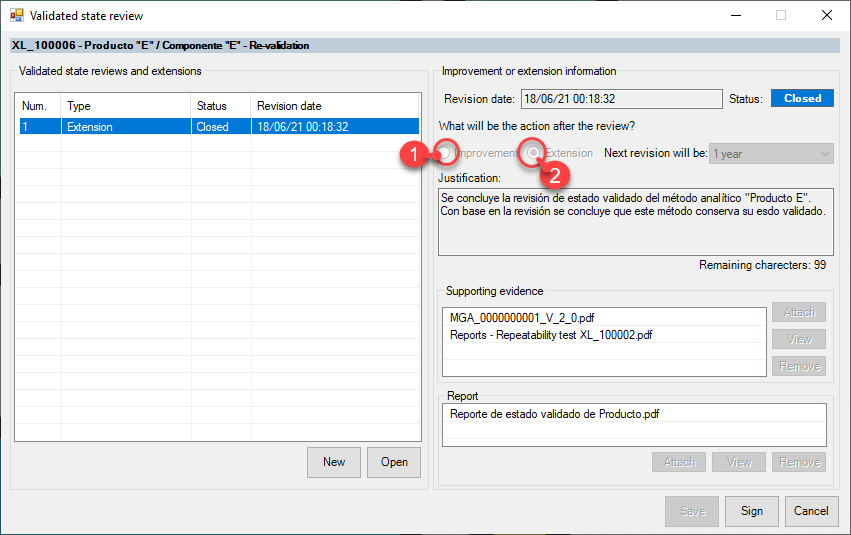
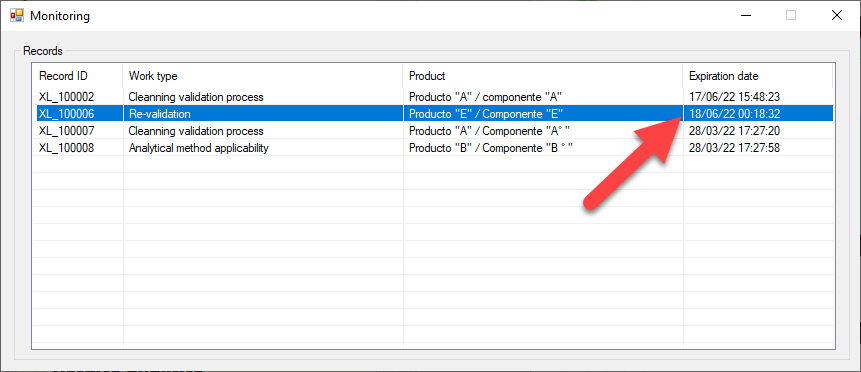
Algunas preguntas relevantes, que le ayudaran a definir el estado de control y/o mantenimiento, son:
- El método fue planteado y validado en concordancia con su categoría?
- La estadística paramétrica es integra?
- Los resultados de validación analítica son consistentes y claros?
- Si es un método compendial, los datos de pruebas de robustez tolerados están documentados?
- Los criterios de “Suitability” del sistema, se mantienen a través de los análisis?
- Las condiciones analíticas establecidas en el método final (preparación , condiciones de análisis, columna etc.) son las mismas que están descritas en el reporte de validación del método?
- Si hay cambios al método, se registran y se alinean a un control de cambio y se emite un CaPa al método y con esto evaluar el impacto a la validación?
- Qué tan recurrentes son las desviaciones o justificaciones asociadas al método analítico?
- Si la validación analítica sigue vigente, puedo hacer un revisión de estado validado antes de su fin expiración si surgen problemas?
No olvides que en caso de ser necesario, puedes contactarnos para mejorar el proceso de laboratorio y administrativo de tu organización a través software 100% confiable y seguro. Recuerda que en deappharma somos expertos en la materia, por lo que procesos complejos no representa un reto para nosotros, debido a que tenemos en mente los aspectos relevantes de cumplimiento.
Referencias.
- Analytical Procedures and Methods Validation for Drugs and Biologics Guidance for Industry
- Pharmaceutical quality system ICH Q10.
- Guidance for Industry Process Validation: General Principles and Practices.
¡Contáctanos! y conoce de que manera te vamos a ayudar.
¡Únete a nuestra comunidad en redes!
7, Jun 2021
La esencia del Límite de Cuantificación en métodos analíticos por HPLC.
Importancia de la validación.
Validación se puede definir como “encontrar o evaluar la verdad de algo”.
Cuando un método analítico es usado para generar resultados acerca de las características de la sustancia de interés (eg., fármaco o medicamento), es vital que los resultados sean confiables, ya que estos serán usados como base para la toma de decisiones en relación, con la definición de la forma de administración del fármaco o medicamento al paciente.
Un estudio de validación, el cual deriva en un método analítico tiene como finalidad el asegurar que los resultados obtenidos sean fidedignos (ALCOA), siempre que estos sean obtenidos dentro del proceso analítico.
¿Pero en donde o porque radica la importancia del proceso de validación?
La importancia radica en que cada día, se realizan en el mundo un alto número de análisis, relacionados al monitoreo de compuestos orgánicos, estas mediciones son utilizadas en algunas situaciones para la toma de decisiones, en áreas como son; control de calidad (liberación), manufactura (evaluación de procesos), áreas analíticas (estudios de estabilidad), proyectos de transferencia analítica.
El límite de Cuantificación “Concepto”
Límite de cuantificación, es la habilidad de un método para detectar y cuantificar la cantidad mínima de un analítico en una muestra, cual puede ser detectada con precisión y exactitud.
El límite de cuantificación es un parámetro de cuantificación de ensayo, para los niveles bajos de los compuestos en las matrices farmacéuticas, y es usada particularmente para la determinación de impurezas y/o productos de degradación.
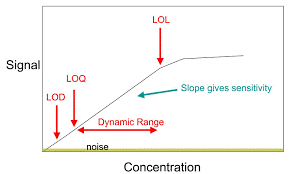
Planteamiento de la prueba.
Se preparan 6 muestras de placebo adicionadas con el componente principal Api (polvo) (Active Pharmaceutical Ingredient) es lo recomendable. Ya que de esta manera podrás retar la esencia de la preparación analítica. Es recurrente contar con una cantidad limitada de impureza, por lo que, se podrá plantear la prueba con un estándar ya disuelto, para posteriormente ser adicionado a las soluciones placebo (n=6) y de esta manera optimizar el uso.
En caso de no contar con el placebo o matriz farmacéutica se podrán preparar 6 muestras independientes de producto terminado caduco o producto que es sometido a pruebas de estrés controlada. La concentración analítica de la muestra estará determinada en relación al 50% ó 0.05% respecto a la concentración establecida, es decir, respecto al límite de sustancias relacionadas, productos de degradación conocidos y/o desconocidos establecidos en tu método de análisis. El restante porcentual será completado con producto terminado, en los que el cálculo de recobro se vera ajustado por la diferencia que habrá entre el nivel evaluado y el blanco (muestra enriquecida) para ajustar la señal de trabajo.
Estadística y parámetros a evaluar (primarios).
- Promedio individual
- Promedio de n=6
- Desviación estándar de n=6
- Mínimo de n=6
- Máximo de n=6
- Intervalos de confianza del valor de “µ”
- T critico
- T estadístico
- Intervalo de confianza de ““µ” %
- P-value
Criterios de aceptación.
La evaluación deberá mostrar las señales del pico de interés diferenciadas de la señal ruido:
- Señal ruido del pico de interés mínimo de 10.
- Los recobros de las 6 muestras deben cumplir con el recobro especificado.
- Debe mostrarse homocedasticidad en los resultados globales n=6, p-value mayor al 0.05
- Los intervalos de confianza deben incluir el valor del 100%.
- El coeficiente de valoración debe ser “<= “al establecido 5, 10, 15 ó 20 % según aplique el nivel % del límite evaluado.
Obtención de la concentración a evaluar.
Las principales formas de obtención del parámetro son:
1.-Basado en evaluación visual: la evaluación visual puede usarse para métodos no instrumentales, pero también puede usarse con métodos instrumentales. el límite de cuantificación se determina mediante el análisis de muestras con concentraciones de analito y estableciendo el nivel mínimo al cual el analito puede ser detectado de manera confiable.
2.-Basado en señal a ruido: este enfoque solo se puede aplicar a los procedimientos analíticos que muestran una línea de base ruido. La determinación de la relación señal / ruido se realiza comparando señales de muestras con bajas concentraciones conocidas de analito con las del blanco muestras y establecer la concentración mínima a la cual el analito puede ser detectado y cuantificado de manera confiable. Generalmente se considera una relación señal / ruido entre o 10: 1 aceptable para estimar el límite de cuantificación.
3.-Basado en la desviación estándar de la respuesta y la pendiente: el límite de cuantificación (QL) puede expresarse como:
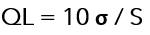
Donde:
σ = desviación estándar de la respuesta.
S = la pendiente de la curva de calibración.
La pendiente S puede estimarse a partir de la curva de calibración del analito.
-Basado en la curva de calibración: se debe estudiar una curva de calibración específica utilizando muestras que contengan un analito en el rango del Límite de Cuantificación. La desviación estándar residual de una línea de regresión o el estándar la desviación de las intersecciones con el eje y de las líneas de regresión pueden usarse como la desviación estándar.
Recomendaciones.
-El límite de cuantificación y el método utilizado para determinar el límite de cuantificación debe ser presentado, el límite debe validarse posteriormente mediante el análisis de un número adecuado de muestras conocidas o preparadas a la concentración del límite de cuantificación.
-La evaluación del parámetro de Limite de Cuantificación siempre se lleva a cabo con enfoque cuantitativo, es decir, tratar el ensayo como si fuese una valoración.
-El recobro no solo se puede ver afectado por la matriz también se ve alterado por la posible adhesión a componentes del contenedor / cierre, por ejemplo, vial de vidrio, válvula dosificadora. En general, un procedimiento de preparación de muestra más simple dará como resultado una menor variación de la recuperación.
No olvides que en caso de ser necesario, puedes contactarnos para mejorar el proceso de laboratorio y administrativo de tu organización a través software 100% confiable y seguro. Recuerda que en deappharma somos expertos en la materia, por lo que procesos complejos no representa un reto para nosotros, debido a que tenemos en mente los aspectos relevantes de cumplimiento.
Proceso de linealidad del sistema y LOQ controlado y gestionado con eADMxL.
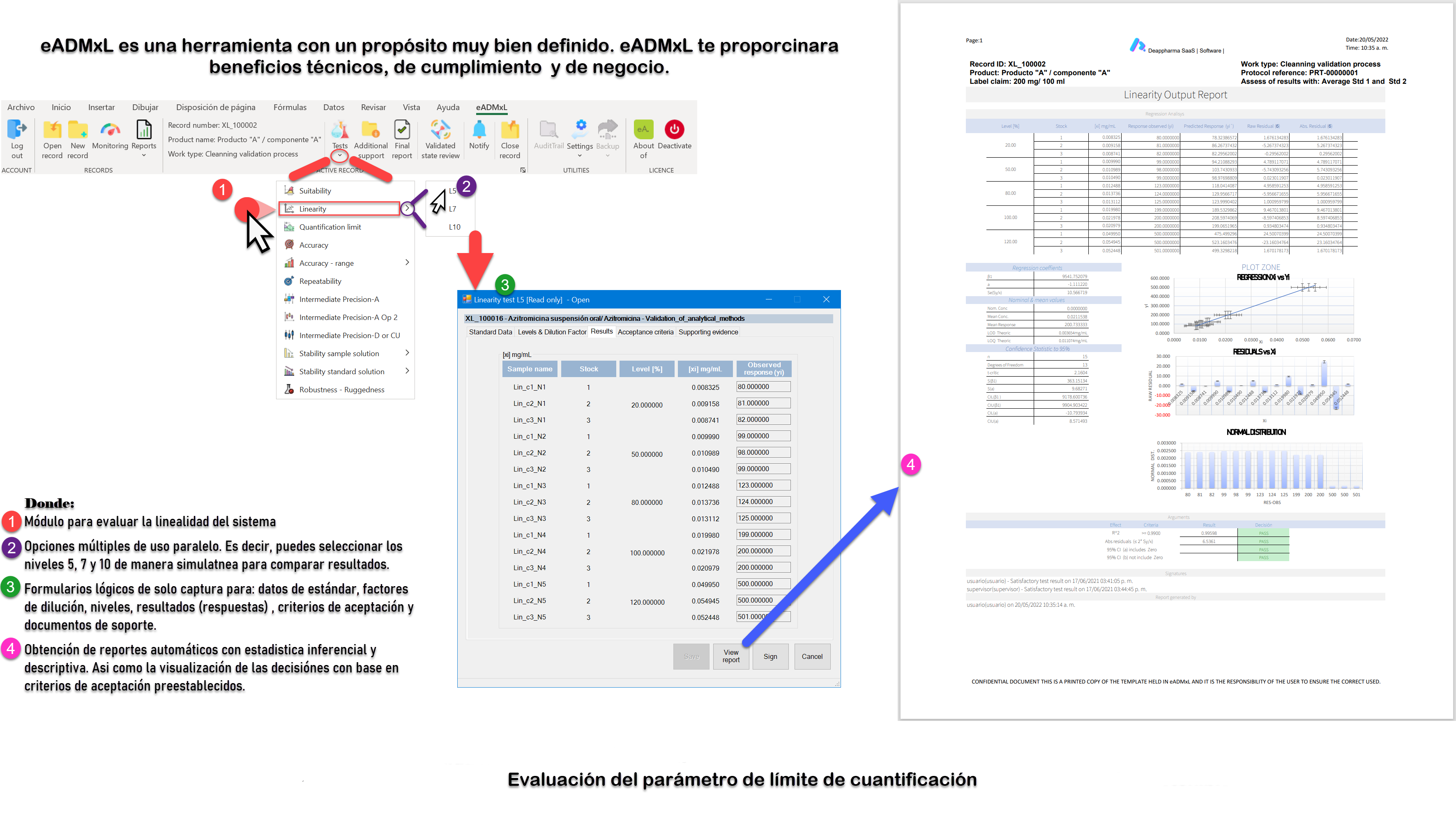
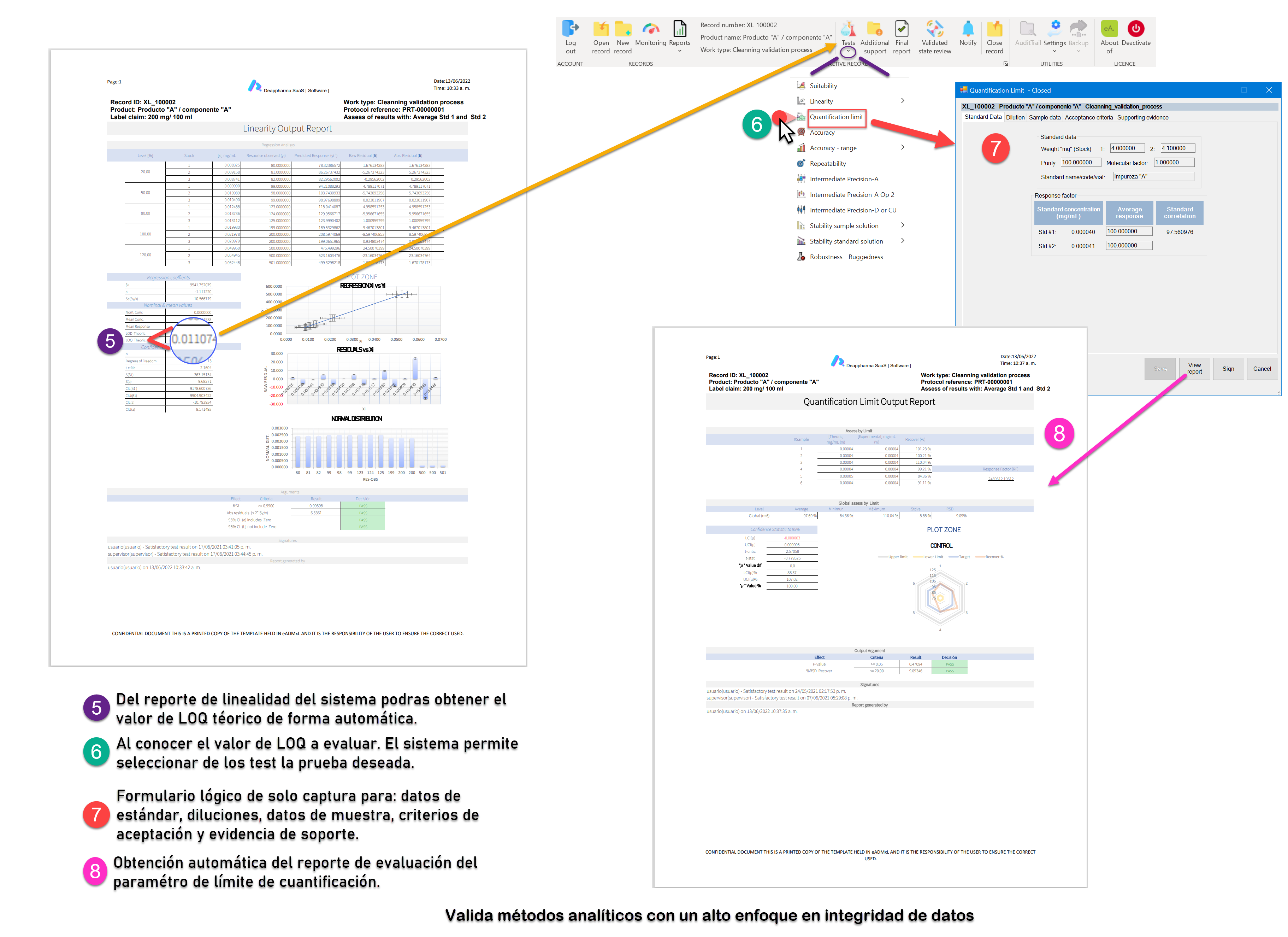
No olvides que, en caso de ser necesario, puedes contactarnos para mejorar el proceso de laboratorio y administrativo de tu organización a través software 100% confiable y seguro. Recuerda que en deappharma contamos con un software que te ayudara a controlar tu proceso de validación de métodos analíticos. Somos expertos en la materia, por lo que procesos complejos no representa un reto para nosotros, debido a que tenemos en mente los aspectos relevantes de cumplimiento.
Referencias.
- NORMA OFICIAL MEXICANA NOM-059-SSA1-2015, Buenas prácticas de fabricación de medicamentos.
- FDA analytical procedures and methods validation for drugs and biologics
- WHO guidelines on validation – appendix 4 analytical method validation
- ICH Harmonised tripartite guideline validation of analytical procedures: text and methodology q2(r1).
¡Contáctanos! y conoce de que manera te vamos a ayudar.
¡Únete a nuestra comunidad en redes!
13, May 2021
Automatización de la gestión documental.
En los últimos años, un gran número de organizaciones ha implementado procesos documentales que evidencian y soportan las funciones realizadas como resultado de sus actividades diarias. Dichos procesos son de carácter secuencial y transversal a las mismas, y están destinados a la planeación, producción, gestión y trámite, organización, transferencia, disposición, preservación y valoración de los documentos, ya sean físicos o electrónicos. Basado en lo anterior, la gestión se realiza, generalmente, a través de la producción de documentos físicos, haciendo uso de planillas de entrega, formatos de préstamos documentales, acuses de recibido, entre otros; y documentos electrónicos como correos electrónicos e información no estructurada que, dependiendo de cada organización, puede ser ejecutada por humanos o por sistemas especializados para tal fin.
En este sentido, una organización produce un extenso número de documentos que, sin una adecuada gestión documental, pueden generar problemas como desperdicio de papel, pérdida de información, gran cantidad de tiempo en consulta o recuperación de información, entre muchos otros inconvenientes administrativos y costes económicos para de la entidad. Para contrarrestar y mitigar este tipo de situaciones, se hace necesaria la implementación de la automatización para la transformación de la gestión documental, entendida como la capacidad de hacer uso de sistemas para llevar a cabo ciertas acciones que normalmente son ejecutadas por personas, soportado con diferentes metodologías y tecnologías.
La automatización de documentos (también conocido como ensamblaje de documentos) es el diseño de sistemas y flujos de trabajo que ayudan en la creación de documentos electrónicos. Estos incluyen sistemas basados en la lógica que utilizan segmentos de texto o información ya existentes para generar un nuevo documento. La automatización de documentos puede ser utilizada para automatizar todo texto condicional, texto variable e información contenidos en un conjunto de documentos.
Los sistemas de automatización permiten a las empresas reducir al mínimo la entrada de datos, el tiempo dedicado a la lectura y corrección, así como los errores humanos. Algunos beneficios adicionales pueden ser: ahorro de tiempo y financiero debido al menor manejo de papel, carga de documentos, almacenamiento, distribución, envío, trabajo y gasto.
En tal razón, la automatización de procesos documentales es un factor clave en la transición de la gestión documental manual a la realizada de forma automática o semiautomática por medio de tecnologías de información.
De esta manera, se puede determinar que la automatización de procesos documentales conlleva una serie de oportunidades para las organizaciones como lo son: reducción de costos, programación automática de tareas, control y seguimiento de estas en tiempo real permitiendo la generación de reportes; disminución de tiempo que deriva en aumento de la productividad y evitando reprocesos por errores operativos. Todo esto, en conformidad con los marcos regulatorios existentes soportados en las políticas “cero papel”, promoviendo la normalización de los archivos en las organizaciones y generando poco a poco una cultura digital.
Beneficios primarios.
Control de versiones.
El auge de los sistemas de gestión documental ha supuesto la mejora y actualización de numerosas funcionalidades. El control de versiones ha estado durante muchos años ligado a la informática favoreciendo la labor de los desarrolladores, pero ¿qué labor puede realizar para mejorar la gestión documental?
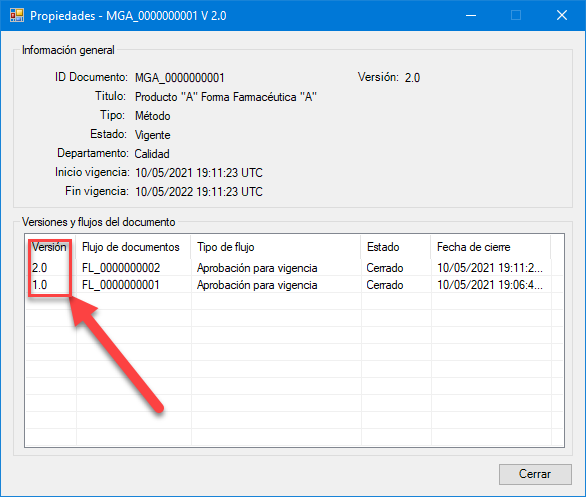
¿Qué es el control de versiones?
Se llama control de versiones a la gestión de cambios efectuados en un documento y otros archivos que contengan información. Los cambios se registran de forma automática y pueden ser identificados mediante números o combinaciones alfanuméricas. En los sistemas de gestión documental con funciones avanzadas de control de versiones, cada cambio no señalará únicamente las modificaciones efectuadas, sino quién y cuándo se realizaron.
Historial de versiones.
El historial de versiones recoge en una misma localización todas las versiones que se han creado de un mismo documento. Es una característica muy diferente dependiendo del tipo de gestor documental con el que se trabaje. Por ejemplo, en los sistemas más avanzados el control de versiones establece la posibilidad de generar un historial ilimitado de versiones al que se podrá volver siempre que se crea necesario.
Seguimiento de cambios.
El seguimiento de cambios es el concepto entorno al que gira la idea del control de versiones. Dependiendo del sistema que se utilice y de lo avanzado de sus características, esta funcionalidad determinará aspectos diferentes.
En los sistemas de gestión documental en los que la colaboración en documentos es un aspecto recurrente, el seguimiento de cambios es una característica fundamental. Por ejemplo, en un documento en el que trabajan diferentes empleados, cada uno aporta parte de sus conocimientos al mismo. Con el seguimiento de cambios no se corre el riesgo de pérdida o modificación de la información, ya que siempre se permite volver a la versión anterior de cualquier archivo.
No olvides que en caso de ser necesario, puedes contactarnos para mejorar el proceso de laboratorio y administrativo de tu organización a través software 100% confiable y seguro. Recuerda que en deappharma somos expertos en la materia, por lo que procesos complejos no representa un reto para nosotros, debido a que tenemos en mente los aspectos relevantes de cumplimiento.
¡Contáctanos! y conoce de que manera te vamos a ayudar.
¡Únete a nuestra comunidad en redes!