31, Ene 2023
Validación de métodos analíticos
Validación en la industria
Validación se puede definir como “encontrar o evaluar la verdad de algo”. Cuando un método analítico es usado para generar resultados acerca de las características de la sustancia de interés (eg., Api o medicamentos), es vital que los resultados sean confiables, ya que estos serán usados como base para la toma de decisiones en relación, con la definición de la forma de administración del fármaco o medicamento al paciente.
Un estudio de validación, el cual deriva en un método analítico tiene como finalidad el asegurar que los resultados obtenidos sean fidedignos (ALCOA ++) siempre que estos, sean obtenidos dentro del proceso analítico.
La validación de métodos analíticos es solo uno de los tipos de validación requerida durante el desarrollo y/o manufactura del producto. Para cumplir con los requerimientos de las buenas prácticas de manufactura (GMP), las compañías farmacéuticas deberán tener una política general de validación, en la cual se detalle que documentación, parámetros y evaluación, serán los necesarios para desempeñar dicha acción, sin dejar de considerar como alcance las validaciones correspondientes a : Procesos de producción, procedimientos de limpieza, métodos analíticos, evaluación y pruebas de control dentro del proceso , así como , los sistemas computarizados.
El objetivo y razón de incluir la validación como un requerimiento en la cGMP y en la NOM 059 (Regulación local), es de asegurar la calidad en cada paso de la fabricación del fármaco o medicamento, y que la calidad no solo sea evaluada bajo un análisis final del producto. La validación está destinada a proveer el aseguramiento de la calidad de un proceso o sistema, y la cual ha sido establecida, diseñada para derivar a la redacción de una metodología de calidad, para ser usada en un análisis de control de; Proceso, liberación y/o estudios de estabilidad.
El aseguramiento de la calidad de un medicamento, no se encuentra solamente por un simple análisis, si no tal y como se mencionó, es el conjunto de análisis con su respectiva y previa documentación, que den soporte de validación en cada etapa del proceso (ALCOA ++ durante toda la cadena de suministro).
Necesidad de la validación
Cada día, se realizan en el mundo un alto número de análisis, relacionados al monitoreo de compuestos orgánicos, y estas mediciones son utilizadas en algunas situaciones para la toma de decisiones, en áreas como son; control de calidad durante el proceso y la manufactura, comercios, y consumo. Entonces, muchas de estas decisiones, están con base en las mediciones o resultados obtenidos, como son; Liberación de un lote, liberación de producto, lanzamiento de un producto, que al final de la cadena de suministro puede afectar la salud, reputación, costos de análisis, retrabajos y un impacto directo económico, si no se evalúa correctamente un dato. Es aquí donde, la evaluación y obtención de un dato reproducible y seguro (Data Integrity) muestra su importancia, por estas razones, los requerimientos de los laboratorios incluyeron el termino de validación de métodos dentro de sus procesos analíticos.
Los governance de concientización han sido establecido como un requerimiento de control de calidad creado por agencias regulatorias para verificar que los laboratorios están correctamente desempeñando este trabajo, grupos de trabajo regulatorio desarrollan guías oficiales para llevar a cabo la validación, así como el establecimiento de sus criterios. Estas referencias (guías) deberían deberán ser evaluadas para garantizar con esto la confianza.
Los métodos analíticos deben ser validados por profesionistas analistas. Un método analítico con poca confianza para evaluar ensayos muy seguramente provea datos falsos, y entonces esto podría desencadenar en un retiro de producto (Recall).
El ejercicio de validación es costoso y consume mucho tiempo. Normalmente esta función es desarrollada por un área de calidad o soporte analítico, lo que puede alterar el trabajo normal del laboratorio. Sin embargo, el uso de métodos validados elimina repeticiones en el laboratorio y mejora el prestigio de laboratorio favoreciendo a los clientes y la rentabilidad a largo plazo.
En resumen, la validación de un método analítico deberá ser desempeñado por las siguientes razones:
- Elevar la integridad y calidad de los resultados.
- Los productos podrán alcanzar la aceptación por agencias internacionales.
- Lograr (cuando aplique) un rango de aprobación como método “oficial/método de referencia” por las agencias regulatorias.
- Te apoya a lograr la acreditación de requerimientos mandatorios para laboratorios por las guías ISO 17025 y requerimientos normativos GxP.
- Mejora el balance financiero del laboratorio.
Propósito de la validación
Un método analítico detalla los pasos necesarios a desempeñar en un análisis, los cuales pueden incluir, pero no limitados a la descripción de; Preparación de las muestras, estándares y reactivos, uso de algún aparato o instrumento, generación de una curva de calibración, uso fórmulas para realizar el cálculo, etc. El propósito de la validación de un método analítico debe ser adecuado, porque de esto depende el diseño hacia el uso previsto para el que será utilizado.
El uso de los métodos analíticos durante etapas del desarrollo del producto y/o manufactura, provee información con relación a lo siguiente y de los cual depende el alcance de validación:
- Potencia, La cual puede estar correlacionada de manera directa a los requerimientos de una dosis conocida o al establecimiento de esta.
- Impurezas y/o sustancias relacionadas, cuales pueden estar relacionados a mostrar el perfil de seguridad impurezas, la interpretación y predicción en la formación de estas (se asocia directamente con el paciente).
- Evaluación de las características claves del fármaco (caracterización), como son; forma cristalina (polimorfismo), liberación del fármaco (forma farmacéutica de dosificación), uniformidad y disolución (pruebas de aptitud de proceso esenciales para pruebas de liberación), las cuales, al ser evaluadas nos dan información sobre las propiedades que pueden comprometer la biodisponibilidad.
- Productos de degradación, los métodos necesitan ser indicativos de estabilidad, siempre y cuando estos sean utilizados en estudios de estabilidad de medicamentos y fármacos (esencial uso de PDA o MS-LC para una correcta evaluación de la pureza cromatográfica y pureza química). Es indispensable que estos productos de degradación estén cuantificados dentro de un umbral adecuado (establecer el límite con base a ICH Q3A (R) e ICH Q3B (R)) que no sea un riesgo para el paciente, las guías anteriormente mencionadas marcan la directriz específicamente para las impurezas que pueden surgir como productos de degradación de la sustancia farmacológica, o que surgen de interacciones entre la sustancia farmacológica y los excipientes o componentes de los materiales de envasado primarios. La directriz establece una justificación para la notificación, identificación y calificación(cuantificación) de tales impurezas, en base a una evaluación científica de las impurezas probables y reales observadas, y de las implicaciones de seguridad, siguiendo los principios elaborados en la directriz principal. Estas guías describen y proponen valores umbral para informar y controlar las impurezas, en función de la dosis diaria máxima de la sustancia farmacológica administrada en el producto.
Los parámetros y efectos descritos anteriormente, así como su buena implementación aseguran que la producción de la sustancia fármaco o medicamento sean consistente.
La validación de métodos analíticos entonces; es un proceso relevante y este debe ser desempeñado. Ya que este proceso genera los datos necesarios para demostrar integridad, confiabilidad y consistencia. Con base en todo el análisis previó, entonces, podemos definir que el propósito de realizar o desempeñar la validación analítica es demostrar que los procesos involucrados en el desarrollo y manufactura del producto, tales cuales son; producción, limpieza y evaluación analítica, pueden ser desempeñadas de una manera efectiva y reproducible y con esto generan calidad, seguridad y eficacia.
Ahora que conoces la la importancia del proceso de validación vamos con el siguiente paso ¡Mejorar el proceso! ¿Quieres saber como?
¡DA CLIC AQUI!
¡Contáctanos! y conoce de que manera te vamos a ayudar.
¡Únete a nuestra comunidad en redes!
- 0
- Por Team deappharma

24, Nov 2022
Registros electrónicos (Identificadores)
Cuando se gestionan documentos o datos de laboratorio dentro de la industria regulada una función importante dentro del entorno de las buenas prácticas de laboratorio en sistemas computarizados e informatizados es aquel relacionado al control de registros. Visto desde una parte de trazabilidad este identificador es la base de búsqueda de algún documento y/o el comienzo para vincular acciones preestablecidas ya que es de carácter único.
Este articulo estará enfocado hacia el proceso de validación de métodos analíticos, sin embargo el concepto general aplica a diferentes esquemas de software eg, sistemas documentales, datos de laboratorio y hojas de cálculo.
Para comenzar con este articulo compartimos las siguientes definiciones, tales cuales anteceden la esencia de estas líneas.
Como definiciones para el proceso de validación de métodos analíticos establecidas por FDA y USP, respectivamente son;
FDA: “La validación de un método analítico es el proceso por el cual se establece, mediante estudios de laboratorio, que las características de desempeño del método cumplen con los requisitos para las aplicaciones analíticas previstas.”
USP: “La validación de métodos, es el proceso por el cual se demuestra que los procedimientos analíticos son aptos para el uso indicado.”
Como se puede percibir en las definiciones anteriores sobre la validación de métodos analíticos, se haca referencia a la palabra “PROCESO”. La definición de proceso se expresa de la siguiente manera: procesamiento o conjunto de operaciones a que se somete una cosa para elaborarla o transformarla.
Como bien sabemos los procesos de validación de métodos analíticos actualmente son realizados con el uso de hojas de cálculo, donde podemos encontrar varios gaps de integridad de datos. Bajo esta situación el control de registros depende de la clave que se asigna al protocolo de validación correspondiente y por ende el sitio donde se almacena la carpeta física con resultados.
Durante nuestra experiencia hemos observado diferentes situaciones desafortunadas para acceder a la información con procesos de gestión actual, los más representativos son:
- La información no se encuentra en el sitio ubicado destinado en el archivo muerto.
- La información impresa se extrajo y nunca se regresó a su sitio.
- La información impresa se puede destruir y generar un reporte nuevo sin posibilidad de trazar la existencia del primer documento.
Derivado de las situaciones anteriores no solo se observa un impacto sobre los documentos y/o datos de laboratorio. Lo que se impacta de manera esencial es la integridad de los datos y la toma de decisiones.
¿Cómo se impacta la integridad?
La integridad se ve impactada por la falta de control sobre datos y registros. Recordemos que la integridad de datos define que los datos y documentos deben ser perdurables para que estos se mantengan: atribuibles, legibles, contemporáneos y exactos. Por ende, la manera actual de gestionar las validaciones carece de integridad con grandes áreas de oportunidad que se deben atender y solucionar.
¿Que son los registros?
Una parte importante para el control de datos es la estructuración sistemática de registros, tales cuales deben ser ordenados y monitorizados. Pero ¿Qué es un registro?
Un registro es toda la información de un elemento que se almacena en un archivo o tabla de base de datos; por ejemplo, el conjunto de datos debe incluir el nombre del producto, tipo de trabajo, dosis, categoría de método a evaluar, tipo de prueba, referencias de protocolos y vigencias. Así como quien y cuando se creó dicho registro. En algunos casos específicos un registro debe permitir la correlación de este con otros previamente realizados.
Como se mencionó los registros son identificadores únicos tales cuales están estructurados por prefijos, sufijos y números establecidos por algoritmos establecidos. De esta manera se da una secuencia definida sin posibilidad de duplicidad y con una completa trazabilidad mediante pistas de auditoría. Es importante considerar que los registros dentro de los sistemas computarizados e informatizados deben ser ordenados e identificados de manera secuencial para una búsqueda eficiente y un acceso rápido.
¿Nosotros como lo hacemos?
A continuación, te mostramos la secuencia de pasos que realiza nuestro sistema estrella para la creación, control y acceso a registros de validación de métodos analíticos. No obstante, te invitamos a revisar nuestros otros desarrollos, tales cuales cuentan también con estas funciones.
Vallamos entonces.
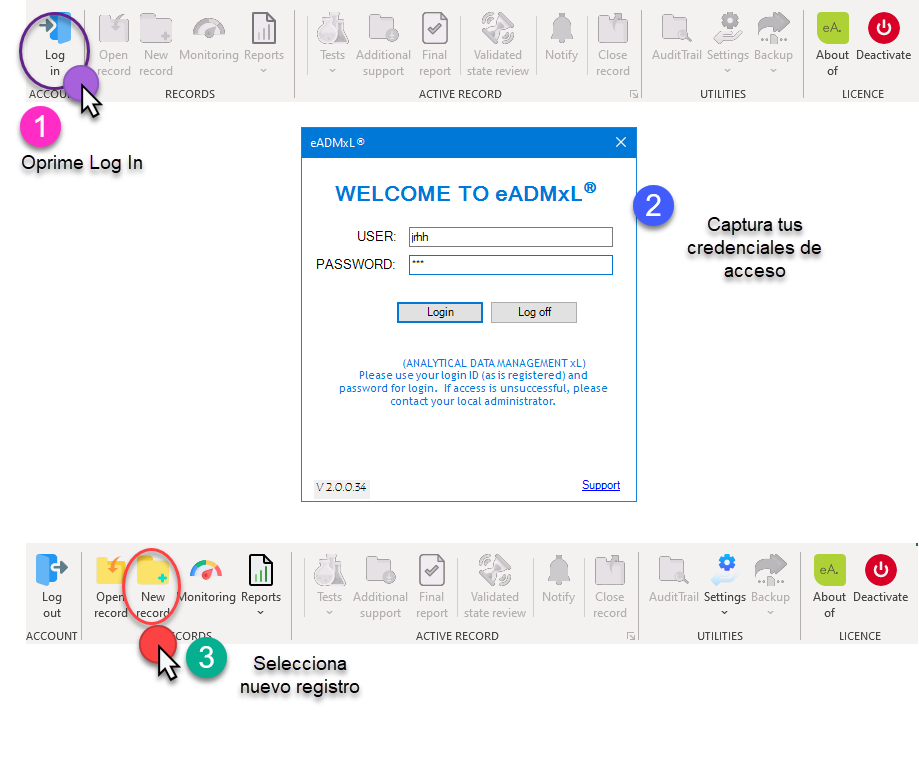
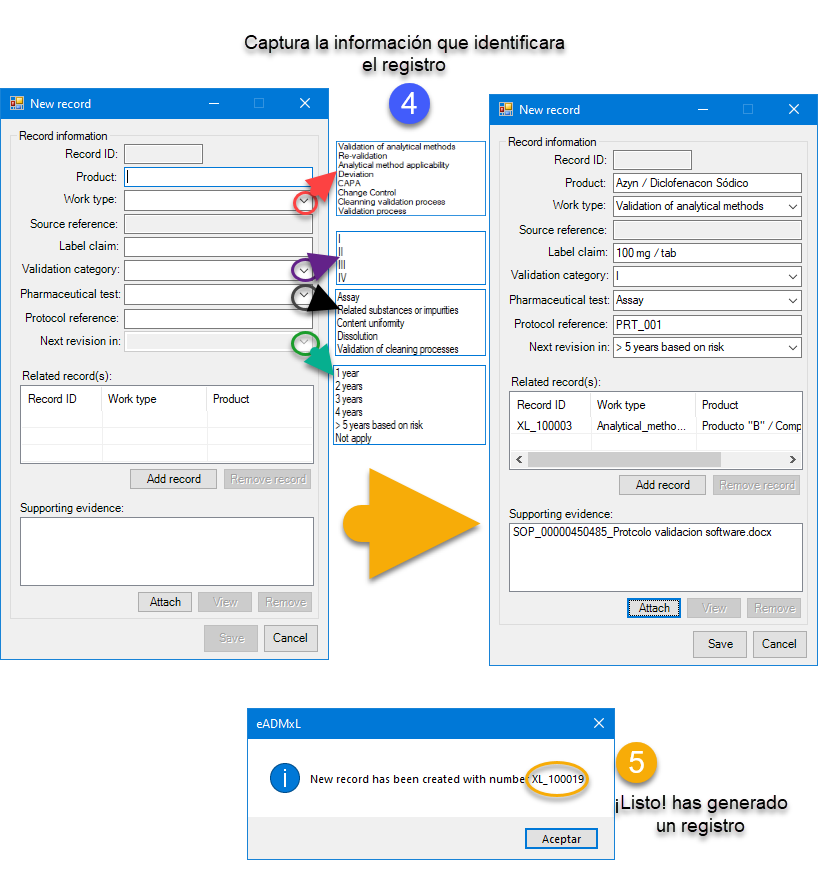
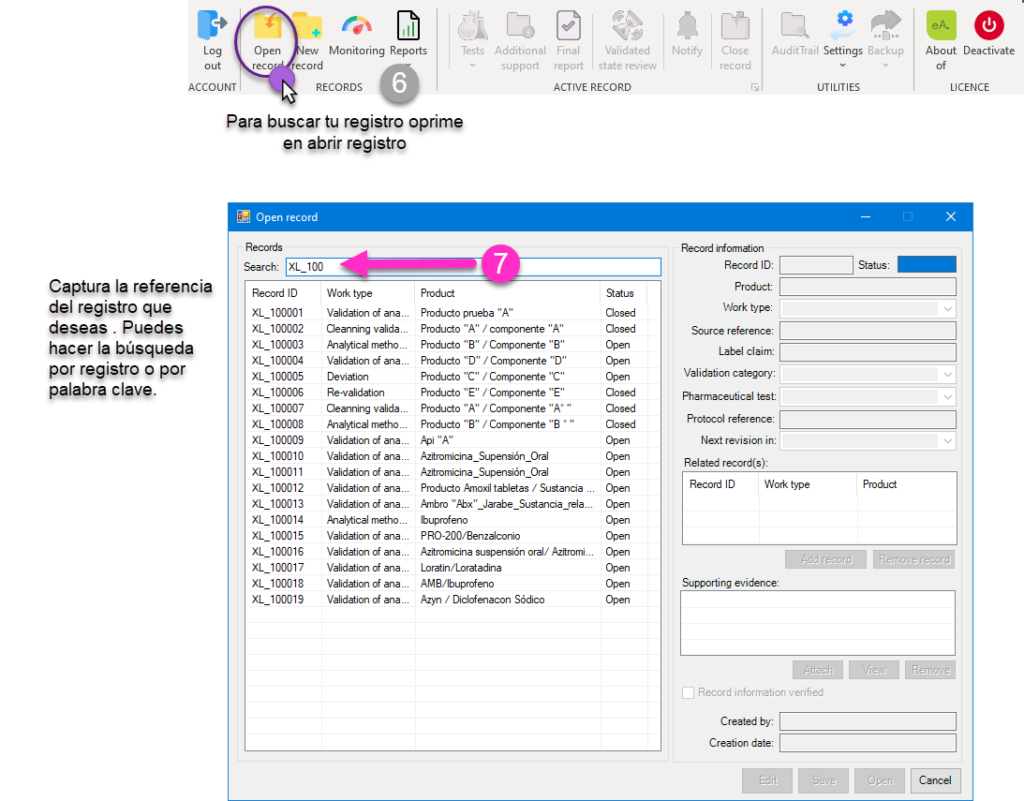
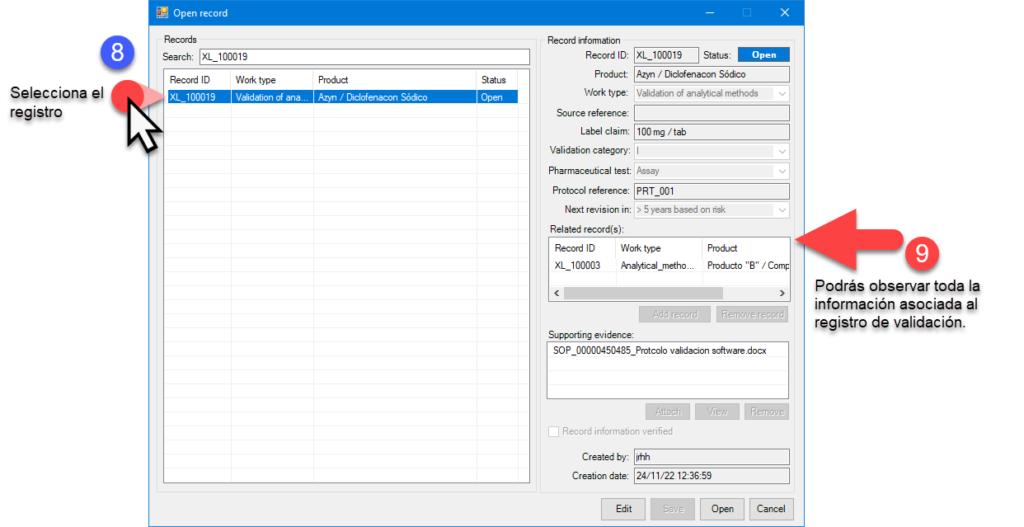
En la actualidad hay muchos gaps relacionados al acceso y control de sistemas computarizados (se incluyen hojas de cálculo). Es por esto que en deappharma desarrollamos herramientas que potencian tu cumplimiento de manera fácil y lógica. Te invitamos a conocer y solicitar tu DEMO y/o implementación de licencia gratuita de nuestro sistema estrella eADMxL para realizar el proceso de validación de métodos analíticos. También te invitamos a explorar nuestros otros desarrollos y visualizar en que proceso te podemos ayudar.
El cambio está en tus manos. Nosotros de ayudamos a potenciar tu cumplimiento.
Referencias:
- FDA draft guidance – Analytical Procedures and Methods Validation
- USP 31 –NF 26, Capítulo General Validación de métodos farmacopéicos
- Villareal de la Garza Sonia, Introducción a la computación. Cap 8 Ed. McGrawHill , México , 2007
¿Necesitas ayuda?
¡Contáctanos! y conoce de que manera te vamos a ayudar.
¡Únete a nuestra comunidad en redes!
10, Nov 2022
¿Por qué es importante el control de accesos en sistemas computarizados en la industria regulada “log in-log out”?
Diariamente dentro de nuestra área de trabajo generamos gran cantidad de “data” que en la mayoría de los casos es de carácter sensible y mucha de esta es utilizada para alguna de las siguientes acciones:
- Toma de decisiones
- Evaluación de datos crudos
- Respuesta ante prevenciones
- Seguimiento a tendencias e históricos entre otros…
Como se bien se sabe los datos que generamos son de carácter GxP y por ende estos deben ser fidedignos y perdurables (esto aplica a Hojas de cálculo y Software). Bajo este hecho, es importante considerar que el acceso a la “Data” debe ser gestionada y monitorizada.
¿Qué es un control de acceso?
Un sistema de control de accesos se puede entender desde una vertiente física. En este sentido, se podría definir como aquel mecanismo que autoriza la entrada al sistema a las personas. El control de acceso informático o control de acceso a sistemas informáticos, en seguridad informática, consiste en la autenticación, autorización de acceso y auditoría. Una definición más estrecha de control de acceso abarcaría únicamente la aprobación de acceso, por lo que el sistema adopta la decisión de conceder o rechazar una solicitud de acceso de un sujeto ya autenticado, sobre la base a lo que el sujeto está autorizado a acceder (perfil de usuario).
Autenticación y control de acceso a menudo se combinan en una sola operación, por lo que el acceso está aprobado sobre la base de la autenticación exitosa.
Es importante mencionar que el control de acceso vinculante está estrechamente ligado a la firma digital ya que este mecanismo utiliza controles de autoridad para permitir firmar o realizar “check” de verificación dentro del sistema sobre las actividades reguladas.
Objetivos de los controles de acceso
El objetivo principal de un control de acceso es salvaguardar la información sobre datos regulados. Es por esto por lo que los sistemas computarizados y software deben contar con las siguientes atribuciones:
- El sistema debe registrar toda creación (nuevos registros y pruebas), modificación y cancelación de autorizaciones de acceso junto con la pista de auditoría asociada.
- El sistema debe utilizar controles de autoridad para garantizar que sólo las personas autorizadas puedan utilizar el sistema, firmar electrónicamente un registro, acceder a la operación o al dispositivo de entrada o salida del sistema informático, alterar un registro o realizar operaciones contemporáneas.
- El sistema debe contar con controles de acceso para garantizar que el personal sólo tenga acceso a funcionalidad que sea apropiada para su papel en el trabajo (perfil de usuario), y que las acciones sean atribuibles a un individuo específico.
- El acceso a la documentación del sistema debe estar restringido a personal autorizado. Un plan de acceso al sistema podría describir estos controles.
- El sistema debe registrar toda creación, modificación y cancelación de autorizaciones de acceso junto con la pista de auditoría asociada.
Se debe considerar que el acceso al sistema debe realizarse a través de credenciales de inicio de sesión individuales utilizando una combinación única de ID de usuario y contraseña, u otros mecanismos de autenticación aprobados. Son aceptables las tecnologías de paso, como el inicio de sesión único, que aprovechan la autenticación anterior del usuario para firmar resultados y/o aprobar reportes.
El control de acceso a sistemas computarizados se realiza con una serie de objetivos puntuales, tales cuales son:
- Restringir o permitir el acceso de personas para sistemas computarizados en determinados procesos.
- Restringir o permitir el acceso a sistemas informáticos, bases de datos y otros servicios de información.
- Proteger los bienes físicos, equipos o datos de las organizaciones ante robos o accesos de terceros sin permiso.
- Detectar accesos no autorizados y poner en marcha mecanismos para evitarlos.
- Registrar y revisar eventos críticos realizados por los usuarios en los sistemas.
- Facilitar la organización de la empresa y el control de los trabajadores.
Principios básicos
Los tres principios básicos que rigen un control de acceso y seguridad son la identificación, la autenticación y la autorización. A continuación, vemos en qué consiste cada uno de ellos.
Identificación: Es el códigoque identifica al trabajador (ID único) y desbloquea el acceso a un sistema computarizado determinado.
Autenticación: En base a estos sistemas se detecta si la persona que está intentando el acceso se encuentra en la base de datos y si cuenta con los permisos necesarios. Es decir, consiste en la verificación de la identidad del usuario.
Autorización: Una vez que el sistema ha identificado y verificado la identidad del usuario, se procede (o no) a autorizar su acceso a los sistemas informáticos.
A estos tres principios se pueden añadir también el de trazabilidad, entendida como el seguimiento o rastreo de productos, personas o datos almacenados en el sistema.
¿El control de acceso forma parte de la validación del sistema computarizado?
El control de acceso forma parte del diseño del sistema. Es por esto que debe ser validado, de tal manera el sistema debe proporcionar la posibilidad de retar los siguientes aspectos:
- Permitir el uso de verificaciones del sistema operativo para hacer cumplir la secuencia permitida de pasos y eventos, según corresponda.
- Las verificaciones de autoridad deberán garantizar que solo las personas autorizadas puedan usar el sistema, acceder a la operación o al dispositivo de entrada o salida del sistema informático, alterar un registro o realizar la operación en cuestión.
- El sistema debe garantizar la unicidad en cada código de identificación de usuario, de modo que no haya dos personas que tengan la misma combinación de código de identificación.
- El sistema debe permitir al usuario autorizado el cambio de contraseña.
- El sistema debe asegurar el registro irrefutable de la identidad de usuarios que ingresan o autorizan datos a través del Audit Trail, el cual deberá registrar todas las acciones que crean, cambien o eliminen registros electrónicos con sus metadatos relacionados.
- El sistema debe generar una auditoria de rastreo segura, generada por el sistema y con sello de tiempo para registrar de forma independiente la fecha y hora de las entradas y acciones del usuario que crean, modifican o eliminan registros electrónicos. Los cambios de registro no ocultarán la información registrada previamente.
En definitiva, los mecanismos de control de acceso son fundamentales para la protección de datos en empresas, y son instrumentos imprescindibles para elevar la seguridad e integridad de los datos en instalaciones, equipos e instrumentos.
¿Nosotros como lo hacemos?
De manera simple te mostramos la secuencia de pasos que realiza nuestro sistema estrella eADMxL. No obstante este comportamiento y cumplimiento esta en cualesquiera de nuestras soluciones.
Te mostramos
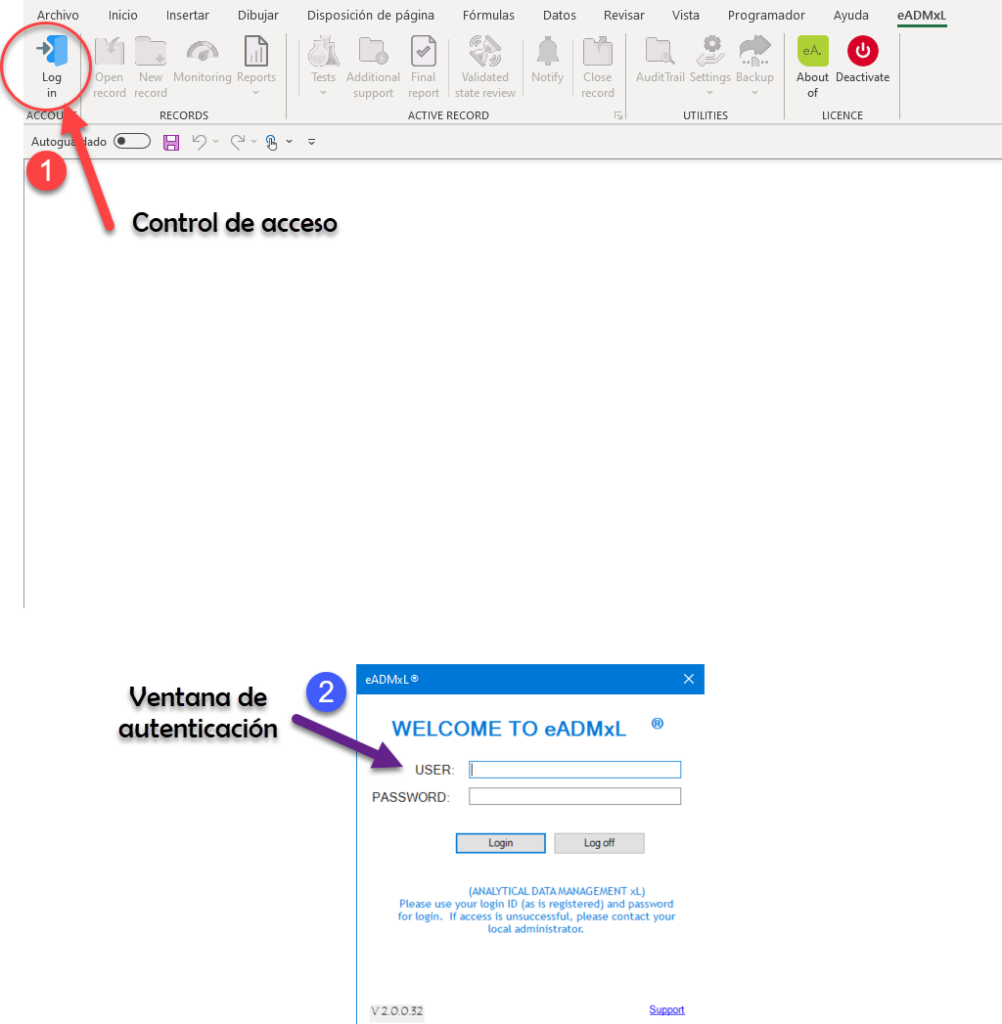
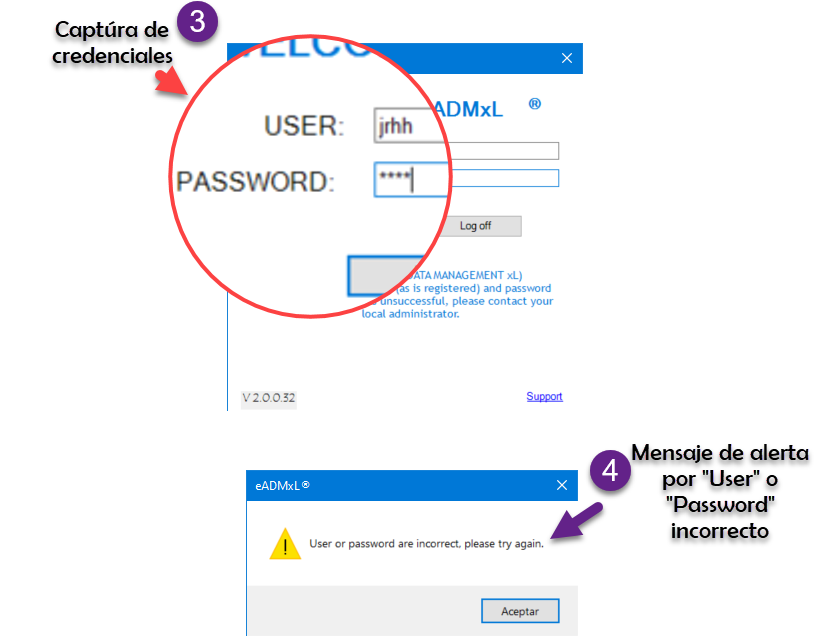
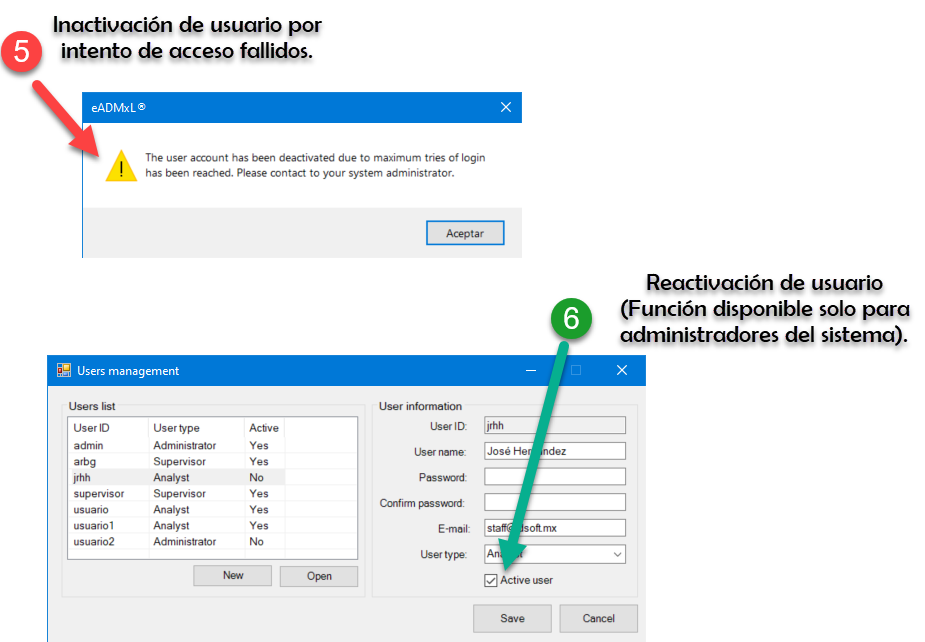
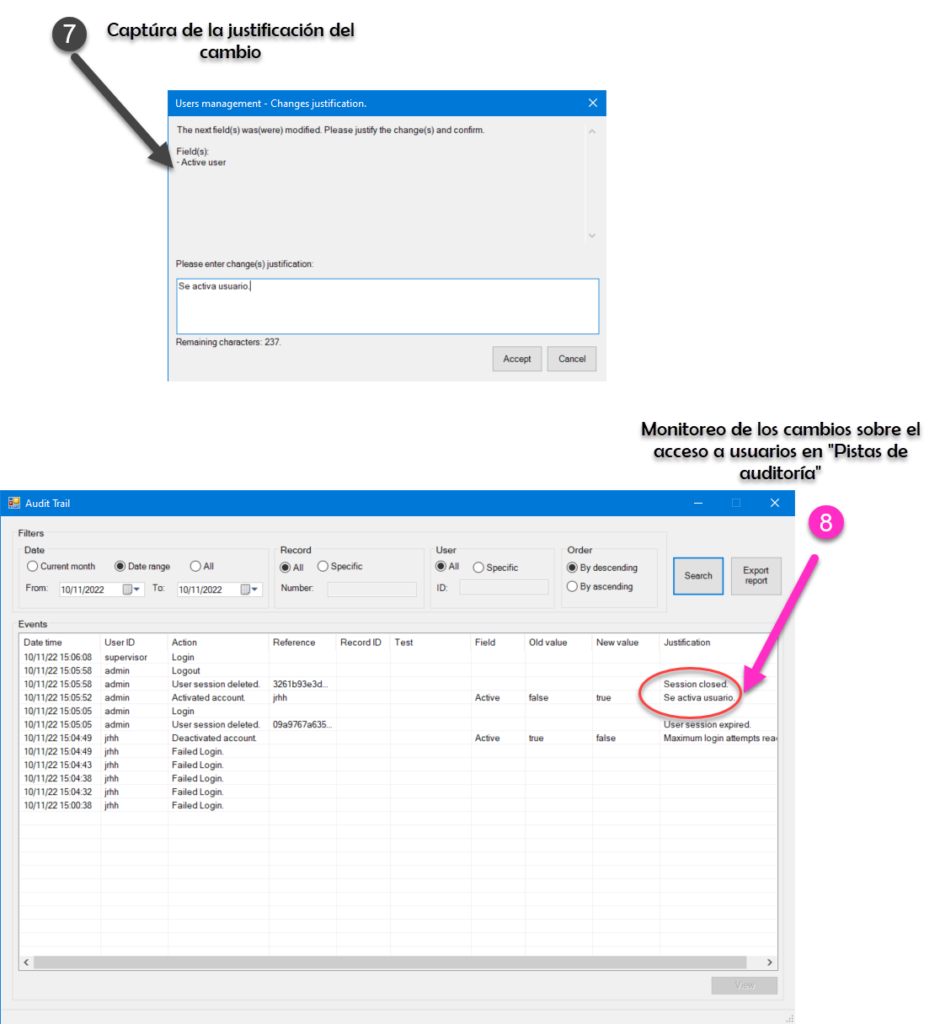
Hacemos impacto que importa
En la actualidad hay muchos gaps relacionados al acceso y control de sistemas computarizados (se incluyen hojas de cálculo). Es por esto por lo que en deappharma desarrollamos herramientas que potencien tu cumplimiento de manera fácil y lógica. Te invitamos a conocer y solicitar tu DEMO y/o licencia gratuita de nuestro sistema estrella eADMxL para realizar el proceso de validación de métodos analíticos y con esto elevar la integridad de los datos de tu laboratorio.
Referencias
Guidance for Industry Part 11, Electronic Records; Electronic Signatures — Scope and Application
GAMP®5: A Risk-Based Approach to Compliant GxP Computerized Systems
Data Integrity and Compliance With CGMP Guidance for Industry FDA
¡Contáctanos! y conoce de que manera te vamos a ayudar.
¡Únete a nuestra comunidad en redes!
15, Sep 2022
Firmas electrónicas (Digitales)
Firmas electrónicas
Las regulaciones del FDA Título 21 CFR Parte 11 establece los criterios sobre los cuales el FDA considera los documentos electrónicos, firmas electrónicas y firmas escritas a mano realizadas a los documentos electrónicos para ser confiables, fiables y generalmente equivalentes a los registros de papel y firmas escritas a mano ejecutadas en papel. Los requisitos generales que establece la FDA son;
(a) Cada firma electrónica deberá ser única para un individuo y no deberá ser reutilizada por, o reasignada a, nadie más.
(b) Antes de que una organización establezca, asigne, certifique o sancione de otro modo la firma electrónica de un individuo, o cualquier elemento de dicha firma electrónica, la organización deberá verificar la identidad del individuo.
(c) Las organizaciones que utilicen firmas electrónicas deberán, antes o en el momento de su utilización, certificar de manera interna que estas están destinadas a ser el equivalente legalmente vinculante de las firmas manuscritas tradicionales.
Componentes y controles de la firma electrónica.
(a) Las firmas electrónicas que no se basen en la biometría deberán
(1) Emplear al menos dos componentes de identificación distintos, como un código de identificación y una contraseña.
Cuando un individuo ejecuta una serie de firmas durante un período único y continuo de acceso controlado al sistema, la primera firma deberá ser ejecutada usando todos los componentes de la firma electrónica; las firmas subsecuentes deberán ser ejecutadas usando por lo menos un componente de la firma electrónica que es solamente ejecutable por, y diseñado para ser usado solamente por, el individuo.
Cuando una persona ejecute una o más firmas que no se realicen durante un período único y continuo de acceso controlado al sistema, cada firma se ejecutará utilizando todos los componentes de la firma electrónica.
(2) Ser utilizadas únicamente por sus auténticos propietarios; y
(3) Ser administrada y ejecutada para asegurar que el intento de uso de la firma electrónica de un individuo por cualquiera que no sea su genuino propietario requiera la colaboración de dos o más individuos.
(b) Las firmas electrónicas basadas en la biometría se diseñarán para garantizar que no puedan ser utilizadas por nadie más que sus auténticos propietarios.
Controles de los códigos de identificación/contraseñas.
Las personas que utilicen firmas electrónicas basadas en el uso de códigos de identificación en combinación con contraseñas deberán emplear controles para garantizar su seguridad e integridad. Dichos controles incluirán:
(a) Mantener la singularidad de cada código de identificación y contraseña combinados, de manera que no haya dos personas que tengan la misma combinación de código de identificación y contraseña.
(b) Garantizar que las emisiones de códigos de identificación y contraseñas se comprueben, retiren o revisen periódicamente (por ejemplo, para cubrir eventos como el envejecimiento de las contraseñas).
Expertos en software comercial y servicios de validación de sistemas computarizados e informatizados
No olvides que en caso de ser necesario, puedes contactarnos para mejorar el proceso de laboratorio y/o administrativo de tu organización a través software 100% confiable y seguro.
Recuerda que en deappharma contamos con software que te ayudara a administrar y controlar procesos especializados. Somos expertos en la materia, por lo que procesos complejos no representa un reto para nosotros, debido a que tenemos en mente los aspectos relevantes de cumplimiento.
¡Contáctanos! y conoce de que manera te vamos a ayudar.
¡Únete a nuestra comunidad en redes!
5, Sep 2022
¿Qué debo saber para abordar la Validación de hojas de cálculo?
Las hojas de cálculo también llamadas planillas, templetes, plantillas de cálculo, son herramientas rutinariamente utilizadas para realizar actividades GxP de carácter normativo. Es por esto que el control y el uso de estas debe ser regulado derivado de que pueden tener impacto sobre la exactitud, fiabilidad, integridad, disponibilidad, autenticidad de los registros y firmas requeridas, según aplique.
Para poder comprender la criticidad e importancia de la emisión de datos con estas herramientas, podríamos partir respondiendo a las siguientes preguntas; ¿Cuál es el objetivo de validar una hoja de cálculo? ¿Qué hace tan viable el uso de las hojas de cálculo como una herramienta en entornos GxP? ¿Qué debo saber antes de comenzar una validación de hoja de cálculo? ¿Cómo definir el alcance de validación de una hoja de cálculo? O más aún ¿Podría auto inspeccionar mi hoja de cálculo? Las respuestas a las preguntas antes mencionadas nos brindan un panorama general de conocimiento.
¿Cuál es el objetivo de validar una hoja de cálculo?
El objetivo de validación de una hoja de cálculo nos encanta definirlo de la siguiente manera: “Buscar la verdad de algo”. Para lo cual debemos revisar, auditar y documentar que una hoja de cálculo emite resultados fidedignos respecto a la especificación de requisitos establecida.
¿Qué hace tan viable el uso de las hojas de cálculo como una herramienta en entornos GxP?
En entornos Gxp se genera una gran cantidad de data que va desde lo simple, lo que podemos denominar texto o escritura hasta data compleja, tendencias, inferencias, decisiones etc. es aquí donde las hojas de cálculo adquieren relevancia ya que estas otorgan al usuario versatilidad, dinamismo y poder de personalización que va desde la vinculación de celdas configurables, hasta la creación de código VBA o MACROS personalizados.
¿Qué debo saber antes de comenzar una validación de hoja de cálculo?
Antes de comenzar una validación de hoja de cálculo es necesario evaluar de manera documentada la hoja de cálculo, es decir definir el riesgo y la expectativa de validación. Una excelente manera para definir es aquella que se relaciona a la complejidad. En función de la complejidad de la hoja de cálculo los resultados requeridos son escalables. Aquí un ejemplo para definir complejidad dentro de este rubro:
- Simple: la hoja de cálculo se considera simple cuando utiliza funciones estándar de Excel. Se pueden utilizar cálculos y otras funciones de Excel (formulas) sencillas siempre que los requisitos estén documentados con suficiente detalle para permitir la realización de pruebas GxP. Si un cálculo o una fórmula no se expresa o no puede expresarse suficientemente en los requisitos, la hoja de cálculo debe considerarse compleja y debe generarse una especificación de diseño.
- Compleja: la hoja de cálculo se considera compleja cuando utiliza funciones no estándar de Excel, como objetos personalizados, macros o Visual Basic. Una hoja también se considera compleja si un cálculo o una fórmula utilizada no se expresa o no pueden expresarse suficientemente en los requisitos indicados anteriormente. Si la hoja de cálculo contiene una amplia codificación personalizada, no debe validarse siguiendo estándares de validación de hojas de cálculo. Es grado de personalización deberá trabajarse como una validación de software utilizado criterios y parámetros de validación diferentes.
¿Cómo definir el alcance de validación de una hoja de cálculo?
El alcance de validación se define con base en la complejidad. Y con base en esta complejidad se define el entregable, un alcance típico es el siguiente:

¿Podría auto inspeccionar mi hoja de cálculo?
La regulación no establece limitantes para que puedas ser tu mismo quien valide tu hoja de cálculo. Sin embargo, se sugiere que la validación sea llevada por un ente ajeno a tu proceso. De esta manera la validación y auditoria de la hoja de cálculo podrá ser realizada de manera objetiva.
Expertos en Validación de Hojas de Cálculo de Microsoft Excel
No olvides que en caso de ser necesario, puedes contactarnos para mejorar el proceso de laboratorio y/o administrativo de tu organización a través software 100% confiable y seguro.
Recuerda que en deappharma contamos con un software que te ayudara a administrar y controlar tu proceso de libros y hojas de Excel. Somos expertos en la materia, por lo que procesos complejos no representa un reto para nosotros, debido a que tenemos en mente los aspectos relevantes de cumplimiento.
Te invitamos a adquirir nuestro servicio de validación, auditoria o diseño de hojas de cálculo y no te pierdas del beneficio extra que tenemos para ti. Conoce este beneficio ¡da clic aquí!
Contáctanos en: info@deappharma.com
¡Únete a nuestra comunidad en redes!
27, Jun 2022
Validación de hojas y libros de Excel
Validación de Hojas de Cálculo de Microsoft Excel
Te has preguntado si validar una hoja de Excel es equivalente a contar con una hoja de Excel integra. Aunque los conceptos parecen a simple vista los mismos. Es una realidad que no. Pero para comprender esto partamos de lo siguiente
Una hoja de cálculo es una estructura tabular de renglones y columnas que permiten realizar operaciones matemáticas, procesar números y ayudar a realizar cálculos desde los más simples hasta los más complejos que implican manipular muchos números y realizar muchas operaciones numéricas.
La importancia de validar una hoja de cálculo Excel reside en que estas pueden afectar directa o indirectamente los atributos de calidad de los productos o procesos. Así como ayudar a una toma de decisión como, por ejemplo: la aprobación de un lote producto para su venta.
La validación de hojas de cálculo Excel no cae fuera de la definición que se establece en la normatividad local e internacional para validación. Existen diversas definiciones de validación, sin embargo, las más completas y consistentes son las de la NOM-059-SSA y la de la FDA, que la definen como:
NOM-059-SSA
<<…”es la evidencia documentada que demuestra que a través de un proceso especifico se obtiene un producto que cumple consistentemente con las especificaciones de calidad establecidas.” …>>
FDA
<<…” el establecimiento de evidencia documentada que proporciona un alto grado de seguridad, que un proceso especifico produce consistentemente sus especificaciones predeterminadas y sus atributos de calidad.” …>>
En donde:
- Documentada. La validación requiere de una minuciosa documentación de todas las acciones que se lleven a cabo dese el inicio hasta el final del estudio.
- Alto grado de seguridad y confianza. Se asume que incluso un programa grande en un sistema complejo computarizado se encuentra realmente libre de errores.
- Proceso especifico. Toda la validación de una hoja de cálculo es un proceso. Por ejemplo, el desarrollo y actividades de prueba de la hoja de cálculo son validadas para asegurar el buen funcionamiento de la hoja de cálculo. Algunas subpartes de validación, tales como calificaciones (instalaciones, operación y desempeño) son especificaciones para cada sistema.
- Consistente. La validación no es un evento de una sola ocasión. El desempeño del sistema de cómputo tiene que ser controlado durante el tiempo de vida del producto.
- Especificaciones. Las actividades de validación comienzan con la definición de especificaciones. El desempeño del sistema de cómputo tiene que ser verificado contra esas especificaciones. El criterio de aceptación está definido por esta prueba.
Enfoque para las hojas de cálculo (Excel) en concordancia con CFR21 parte 11 como sistema cerrado
Las hojas de cálculo Excel son considerados registros electrónicos, por este hecho deben ser tratadas como un sistema informatizado.
Un registro electrónico considera, los documentos y registros que son creados, modificados, mantenidos, archivados, recuperados y/o transmitidos a través de sistemas electrónicos.
Cuando se utilicen sistemas electrónicos para la creación, modificación, mantenimiento, archivo, recuperación y/o transmisión de registros electrónicos deberán establecer procedimientos y controles diseñados para asegurar la autenticidad, integridad y cuando aplique confidencialidad de los registros electrónicos y para asegurar que las firmas electrónicas no puedan ser declaradas como no genuinas. Los procedimientos y controles deben incluir:
- La validación de los sistemas para asegurar la exactitud, confiabilidad, funcionalidad, consistencia y la habilidad para distinguir entre registros inválidos o alterados.
- La habilidad de los sistemas o aplicaciones computacionales para generar copias de los registros exactas y completas, legibles tanto en su versión manual como electrónica, que permitan su inspección, revisión y copia.
- La protección de los registros, que permita su recuperación en forma rápida y exacta durante todo el periodo de conservación de estos.
- El permitir el acceso al sistema únicamente a personas autorizadas.
- El uso de los procesos de auditoría de rastreo seguros, generados por computadora, para registrar en forma independiente el acceso al sistema, así como las acciones que creen, modifiquen o borren registros electrónicos.
- Cotejos operacionales del sistema para obligar que los pasos y eventos ocurran en la secuencia establecida.
- Los cotejos para asegurar que solamente personas autorizadas puedan utilizar el sistema, firmar electrónicamente un registro, acceder a la operación del dispositivo de entrada y salida del sistema computarizado, modificar un registro o realizar la operación manual.
- La determinación de que las personas que desarrollan mantienen o utilizan sistemas de firmas/registros electrónicos tienen la capacidad, adiestramiento y experiencia para llevar a cabo sus tareas.
Firmas electrónicas
Las firmas electrónicas son una compilación de datos en la computadora ejecutados para algunos símbolos, adoptados o autorizados por un individuo que son equivalentes a su firma manuscrita. Para el caso de firmas electrónicas se debe considerar:
- Debe contener la información asociada con la firma que claramente indiquen el nombre en letra de molde de la persona que forma, la fecha y hora de cuando fue ejecutada la firma y el propósito asociado con la misma.
- Estas deben ser únicas para cada persona y cuando se dé el caso de un cambio, esta no debe repetirse o reasignarse a otra persona.
- Cuando el uso de las firmas electrónicas sea adoptado, se debe establecer la fecha a partir de la cual las firmas electrónicas son vigentes y equivalentes a las firmas en manuscrito, para lo cual es necesaria una certificación en un forma u hoja de papel y firmada con una firma en manuscrito.
Que debemos considerar al validar hojas de cálculo (Excel)
Como hemos descrito anteriormente las hojas de cálculo de Excel son consideradas sistemas computarizados. ¿Pero que hay acerca del proceso de validación?
Antes de comenzar debemos conocer y/o actualizar el plan maestro de validación, ya que en este documento deberemos describir por lo menos lo siguiente:
- Nombre del archivo de la hoja de cálculo incluyendo el número de versión
- Ubicación y lugar de almacenamiento de la hoja de cálculo
- Sistema operativo y Software utilizado (resumen de las instalaciones, sistemas, equipos y procesos a evaluar.
Para comenzar debemos partir de dos supuestos que serán los que necesitamos para alcanzar el objetivo. Obtener certidumbre sobre la integridad de datos de las hojas de cálculo Excel es un proceso que se debe llevar de manera sinérgica. Con esto el control y trazabilidad se otorga mediante soluciones informáticas que eleven sustancialmente el control y administración de nuestros libros de Excel que están validados y auditados de manera adecuada. Compartimos contigo lo siguiente para llevar a cabo el proceso de calificación/validación de libros de Excel.
Antes de iniciar con la validación de la hoja de cálculo de Excel debemos establecer los requerimientos de usuario de dicho libro. Recuerda que los requerimientos de usuario establecen de manera generar el alcance que tendrá el libro, sin describir de manera detallada el proceso de diseño y funcionalidad inicial.
Una vez establecidos los requerimientos de usuario del libro de Excel, podemos comenzar con el diseño de la hoja. De tal manera que desarrollemos la estructura de esta lo más acorde a lo establecido.
No obstante, también es factible desarrollar primero el libro y después estructurar los requerimientos de usuario. Esta opción favorece la redacción del documento de especificación.
El diseño de la hoja es una de las etapas más sensibles, ya que el formato y vinculación de cálculos se establece de manera sistemática y con escrutinio. Sin embargo, la verificación, validación y auditoria debe hacerse por un área ajena no dueña del Libro (recomendable).
Una vez se cuente con el desarrollo y diseño del libro de Excel viene la tarea de calificar/validar y auditar el archivo. Las consideraciones que deben formar parte de la documentación de soporte de validación son:
- Evaluación de precedencias. Esta evaluación indica el orden en que se ejecutan los cálculos en una fórmula si esta contiene varios operadores.
- Evaluación de celdas precedentes y celdas dependientes. Las celdas precedentes son aquellas que se refieren a fórmulas de otras celdas. Por otro lado, las celdas dependientes son aquellas que contienen fórmulas que se refieren a otras celdas.
- Referencias circulares. Este término se refiere al hecho de que una fórmula utilice la celda que lo contine como uno de sus parámetros de forma directa o indirectamente.
- Control de cálculo. Por defecto, Excel recalcula toda la hoja de cálculo cuando introduce un cambio en alguna de sus celdas.
- Inspección. La ventana de inspección es una función que forma parte de la auditoria de fórmulas de Excel. Con ella es posible visualizar la fórmula o las fórmulas de una misma hoja de cálculo de modo completo con el fin de facilitar su revisión, control y confirmación.
- Comprobación de errores. Se utiliza para localizar e identificar errores que pueden cometerse al introducir fórmulas.
- Revisión de ortografía. Se utiliza para corregir los textos contenidos dentro de los textos existentes en la hoja de Excel.
- Protección de la hoja. Existen diferentes herramientas en Excel para impedir que un usuario modifique, mueva o elimine accidental o premeditadamente los datos contenidos en las celdas de una hoja de cálculo o libro.
- Protección de la estructura del libro. Esta función impide la modificación de toda la estructura del libro, es decir, no permite mover, eliminar o agregar hojas, modificar su nombre, cambiar el tamaño de las ventanas, etc.
- Conservación. Esta función permite migrar un libro u hoja de Excel respectivamente hacia un sistema de alto control para su administración.
- Acceso seguro a libros de Excel. Esta función permite acceder de manera controlada al libro de Excel.
- Control de versiones. Esta función permite contralar mediante un explorador virtual las versiones de libros y hojas de cálculo.
- Pistas de Auditoria. Esta función permite establecer mediante un control de programación la estructuración dinámica de un “Audit Trail” sobre uso de cierto libro de Excel y su versión.
- Respaldo y Restauración. Esta función permite establecer la posibilidad de respaldar todos los archivos (libros de Excel) contenidos en la carpeta virtual.
Como lo has podido percibir, el alcanzar una hoja o libro de Excel con atributos de integridad de datos se vincula de estrechamente entre los controles “per se” de Excel y la sinergia con software de gestión y control.
Recuerda que en deappharma contamos con una herramienta que te ayudara a administrar y controlar atributos de tus hojas y/o libros de Excel para obtener un optimo desempeño de integridad y cumplimiento.
Beneficios de la validación de hojas de cálculo
Validar, administrar y controlar las hojas de cálculo o libros de Excel permite a las organizaciones:
- Permite mitigar uno de los riesgos más significativos en la industria. Siendo este la pobre integridad de las hojas de cálculo y su control.
- Los recursos físicos son optimizados como consecuencia del protagonismo digital. Así, el escaneo de los documentos físicos hace posible una reducción del tamaño de las instalaciones reservadas al archivo y custodia de todos los libros de Excel de la compañía.
- Centralización de la información, gracias a la cual se potencia tanto el flujo de trabajo como la colaboración entre distintos departamentos y, algo muy importante, entre distintas sedes o lugares de trabajo separados entre sí.
- Mayor grado de control de los diferentes libros gracias a la monitorización del ciclo de vida del archivo.
- Mayor seguridad, pues las copias de seguridad evitarán la destrucción de los libros, así como también se puede llevar un control sobre quién accede a los distintos libros y quién modifica la estructura de estos. Por lo que la sustracción de información sensible puede ser fácilmente rastreable.
Expertos en Validación de Hojas de Cálculo de Microsoft Excel
No olvides que, en caso de ser necesario, puedes contactarnos para mejorar el proceso de laboratorio y administrativo de tu organización a través software 100% confiable y seguro. Recuerda que en deappharma contamos con un software que te ayudara a administrar y controlar tu proceso de libros y hojas de Excel. Somos expertos en la materia, por lo que procesos complejos no representa un reto para nosotros, debido a que tenemos en mente los aspectos relevantes de cumplimiento.
Al mandar con nosotros tus libros y Hojas de Excel para validar, diseñar o auditar obtendrás nuestro software eDocuSeed de manera gratuita por un año. Conoce los beneficios de nuestro aplicativo ¡Da clic aquí!.
También te pueden interesar los siguientes artículos ¡Da clic en alguno de ellos!
Categorización de hojas de cálculo
Tipificación de hojas de cálculo
¡Contáctanos! y conoce de que manera te vamos a ayudar.
¡Únete a nuestra comunidad en redes!
15, May 2022
Idoneidad del sistema (HPLC, MS-HPLC)
Las pruebas de idoneidad del sistema son una parte integral de muchos procedimientos analíticos. Las pruebas se basan en el concepto de que el equipo, la electrónica, las operaciones analíticas y las muestras a analizar constituyen un sistema integral que puede ser evaluado como tal. Los parámetros de las pruebas de idoneidad del sistema que deben establecerse para un procedimiento concreto dependen del tipo de procedimiento que se valide.
La evaluación de la aptitud del sistema se recomienda para todos los métodos analíticos, ya que permite verificar que el sistema de medición funciona apropiadamente, independientemente de las condiciones ambientales. Es conveniente que antes de llevar a cabo la validación del método analítico, se establezcan los criterios apropiados para la operación del sistema de medición, para ser evaluados en la validación y verificados de manera rutinaria al emplear el método analítico.
Métodos cromatográficos
La solución de aptitud del sistema se establece en el desarrollo del método y puede ser una solución del analito al 100 % del rango de linealidad o más concentrado. Puede ser una mezcla del analito con otro componente para verificar la resolución del analito y su(s) compuesto(s) de degradación o relacionados (ya sea agregándolos o degradando la muestra).
Metodología
La evaluación se recomienda con la inyección por sextuplicado la solución de aptitud del sistema. Calcular el Coeficiente de Variación (CV) del analito y se procede reportar.
- Factor de capacidad (K’)
- Resolución (R)
- Tailing ( T )
- Platos teóricos (N)
- Factor de coleo
Los valores mínimos y/o máximos para cada uno de los parámetros anteriores, son los siguientes:
- Factor de capacidad (K’) > 10 (recomendable)
- Coeficiente de variación (CV) ≤ 2% para proceso de valoración, disolución y ≤ 20% para procesos de evaluación de impurezas individuales.
- Resolución (R) > 1.5 (recomendable si se observa la presencia de productos de degradación y/o impurezas)
- Tailing ( T ) <1>
- Factor de coleo >2.5
Actualmente los procesos y evaluación de métodos analíticos por cromatografía liquida (HPLC, MS-HPLC etc..) establecen como parámetro de evaluación la idoneidad del sistema. Sin embargo como proceso integral de la validación de métodos analíticos esta prueba debe formar parte evaluada de este. De esta manera podemos obtener de manera verificable el inicio de las condiciones de validación.
Las pruebas de idoneidad del sistema son pruebas que deben ser evaluadas previamente a lanzar alguna corrida analítica, con el objetivo de verificar que las condiciones del sistema son optimas para llevar a cabo el análisis. La idoneidad del sistema siempre debe ser evaluada con soluciones de referencias, es decir estándares. No es sugerible que la idoneidad incluya la inyección y previsualización de resultados (áreas) de muestras. Ya que esta acción de inyección de muestras previamente a lanzar la corrida analítica, son consideradas actualmente malas practicas de laboratorio en lo asociado a integridad de datos.
Recordemos que la idoneidad del sistema debe formar parte integral del proceso de validación de manera inicial ya que esta es una buena referncias para determinar el momento de fallo y establecer limites de especificaciones vinculadas a este parametro de desempeño.
Proceso de idoneidad controlado y gestionado con eADMxL.
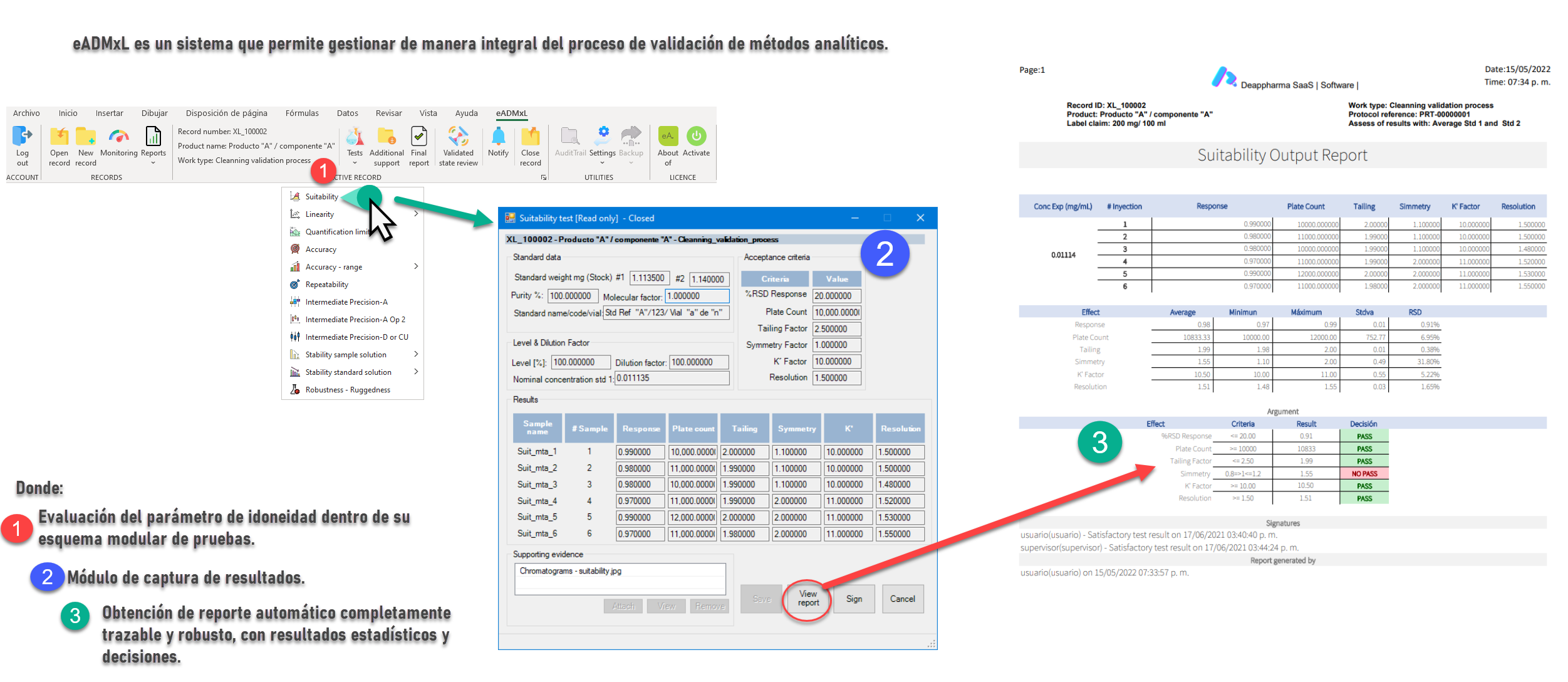
No olvides que, en caso de ser necesario, puedes contactarnos para mejorar el proceso de laboratorio y administrativo de tu organización a través software 100% confiable y seguro. Recuerda que en deappharma contamos con un software que te ayudara a controlar tu proceso de validación de métodos analíticos. Somos expertos en la materia, por lo que procesos complejos no representa un reto para nosotros, debido a que tenemos en mente los aspectos relevantes de cumplimiento.
Referencias
- VALIDATION OF ANALYTICAL PROCEDURES: TEXT AND METHODOLOGY Q2(R1)
¡Contáctanos! y conoce de que manera te vamos a ayudar.
¡Únete a nuestra comunidad en redes!
11, Feb 2022
Audit Trail para asegurar y elevar el proceso de Integridad de datos
Este término regulatorio e informático se refiere a la auditoría, detección continua y exhaustiva de los datos. Por lo que es relevante contar con este proceso o función en cada uno de nuestros sistemas informatizados. El objetivo, es trazar la vida de un dato de manera perdurable.
La industria farmacéutica, dispositivos médicos y almacenes utilizan sistemas informatizados dentro de sus procesos. Por este hecho, es relevante el control y trazabilidad de datos. Para abordar el tema y conocer el alcance e importancia de la función de pistas de auditoria te invitamos a leer lo siguiente.
¿Qué es un “pista de auditoría”?
La «Pista de auditoría» o también conocida como «Audit Trail». Significa un registro electrónico seguro, generado por computadora con sellos de tiempo que permiten la reconstrucción del curso de los eventos relacionados con la creación, modificación y/o eliminación de un registro electrónico. Por ejemplo, la pista de auditoría para una ejecución de cálculos analíticos con la utilización de Excel como software de cálculo, debe incluir como mínimo lo siguiente: nombre de usuario, la fecha/hora de la ejecución, los cambios realizados sobre un valor y los detalles justificados de dicho cambio, según apliquen. ¡Pero no solo eso! Las pistas de auditoria son un aparte esencial de los sistemas computarizados. Es por esto, que los reportes de auditorita deben ser robustos y seguros.
Retomemos el ejemplo de uso de la hoja de Excel para obtención de resultados analíticos. En la actualidad la industria configura la obtención de pistas de auditoría sobre los mismos libros de Excel. Esta pista «LOG» regularmente la establecen dentro del mismo libro de manera anidada como una pestaña (hoja) de cálculo. ¡Esta práctica, es poco sostenible y segura! ya que la trazabilidad la dejas en manos de Excel y de la vulnerabilidad de código realizado con VBA.
¿Existe riesgo de perder la trazabilidad en mis libros de Excel por usar configuración VBA?
La respuesta es ¡Si! y tu impacto será alto porque no tendrás la manera de trazar la información.
Los riesgos los podemos clasificar en dos escenarios posibles:
- Daño en el archivo de Excel que no permita acceder a la información.
- Alteración y vulnerabilidad de la programación en VBA (Hasta videos en youtube te enseñan a desbloquear código relizadoo con VBA. ¡Ojo con esto!)
¿Quién debe revisar los registros de auditoría?
La revisión de la pista de auditoría es similar a la evaluación de las tachaduras en papel al revisar los datos. Personal responsable de la revisión de registros bajo cGMP debe revisar las pistas de auditoría que capturan los cambios en datos asociados con el registro a medida que revisan el resto del registro. Para ejemplo, todos los registros de producción y control, que incluyen registros de auditoría, deben revisarse y aprobado por la unidad de calidad. Las regulaciones brindan flexibilidad para tener algunas actividades revisadas por una persona que supervisa o verifica directamente la información.
FDA recomienda un enfoque de sistema de calidad para implementar la supervisión y revisión de cGMP registros.
¿Con qué frecuencia se deben revisar los registros de auditoría?
Si la frecuencia de revisión de los datos se especifica en las regulaciones de cGMP, adhiérase a esa frecuencia para la revisión de la pista de auditoría. Por ejemplo, requiere revisión después de cada paso significativo en fabricación, procesamiento, empaque o almacenamiento, y requiere la revisión de datos antes del lote liberar. En estos casos, aplicaría la misma frecuencia de revisión para la pista de auditoría.
Si la frecuencia de revisión de los datos no está especificada en las regulaciones de cGMP, debe determinar la frecuencia de revisión de la pista de auditoría utilizando el conocimiento de sus procesos y la evaluación de riesgos.
La evaluación de riesgos debe incluir la evaluación de la criticidad de los datos, los mecanismos de control e impacto en la calidad del producto. Los riesgos para los datos incluyen, entre otros, la posibilidad de que se eliminen modifique o excluyan sin autorización o sin detección.
Su enfoque para la revisión de la pista de auditoría y la frecuencia con la que la realiza deben garantizar que se cumplen los requisitos de cGMP, se implementan los controles apropiados y la confiabilidad de la revisión.
¿Puedo aumentar la trazabilidad de mis libros de Excel sin necesidad de usar VBA?
¡Si! y todo de manera virtual con el suso de nuestro complemento COM. Esto garantiza que tus datos sean trazados de manera perdurable.
En deappharma hemos desarrollado eDocuSeed una herramienta validada y especializada para el mantenimiento, gestión y trazabilidad de libros de Excel. ¡Sin duda te ayudaremos a mitigar tu riesgo! Puedes contactarnos para mejorar el proceso administrativo de tu organización a través software 100% confiable y seguro.
Referencias
- Data Integrity and Compliance With Drug CGMP Questions and Answers Guidance for Industry
- Guía para el enfoque de sistemas de calidad de la industria para las regulaciones CGMP farmacéuticas.
¡Contáctanos! y conoce de que manera te vamos a ayudar.
¡Únete a nuestra comunidad en redes!
20, Dic 2021
Hojas de cálculo con integridad de datos con cumplimiento CFR21
Las industrias reguladas utilizan las hojas de cálculo para muchos propósitos. Haciendo que Excel cubra diferentes necesidades atribuidas a las actividades de rutina. Este programa «Excel» puede: calcular, organizar y analizar datos, de tal manera que pueda evaluar el funcionamiento de: procesos, métodos, tendencias y en algunas ocasiones la obtención de documentos, con base en plantillas que se alimentan de bases de datos creadas propiamente en la misma aplicación. Las hojas de cálculo se configuran en un formato de cuadrícula. Cada pieza de información se coloca en su propia celda para que pueda ser usada adecuadamente. Los datos pueden ser números o palabras. Las palabras se organizan alfabéticamente y los números numéricamente. Los números se calculan con suma, resta, multiplicación, división y en algunos casos estadística más avanzada. Partiendo de esto , el uso de hojas de cálculo dentro de la industria regulada toma relevancia, ya que impacta en mayor medida sobre procesos de carácter GxP.
La situación actual
La versatilidad de las hojas de cálculo proporciona opciones de diferentes alcances GxP, por ejemplo;
Bases de datos, con el siguiente alcance:
- Control de muestras
- Control de resultados de resultados de laboratorio
- Listado de muestreos de materiales
- Listado de muestra de retención
- Listado de estatus de documento
- Asignación de código de documentos
- Listados maestros de estabilidades
- Listados de toma de muestras para programa anual, entre otros.
Hojas de cálculo, con el siguiente alcance:
- Emisión de resultados de: ensayo, uniformidad de contenido, sustancias relacionadas, disolución
- Evaluación de todos parámetros de desempeño del proceso de validación de métodos analíticos
- Evaluación de resultados de perfiles de disolución, entre otros.
Plantillas, con el siguiente alcance:
- Emisión de certificados de análisis
- Emisión de protocolos
- Emisión de reportes, entre otros.
Derivado del dinamismo y la versatilidad de la aplicación Excel, podemos obtener diferentes magnitudes de hallazgos relacionadas a eventos de integridad de datos, tales cuales ponen en cuestionamiento el cumplimiento regulatorio.
Las situaciones relevantes por el uso de hojas de cálculo son los siguientes:
Desarrollo
- Desarrollo de plantillas con esquemas de reedición
- Desarrollo de plantillas en los que no ha sido retada la estadística contenida en estas
- Desarrollo de fórmulas estadísticas establecidas, sin una base de conocimiento estadístico.
Funcionalidad
- No cuentan con perfiles robustos para el ingreso a la plantilla, niveles de usuario y definición del alcance de estos para el desempeño en el uso de mismas
- No hay validación de las celdas, discriminando si son numéricas o textuales
- Seguridad de las hojas solo estableciendo contraseñas con las propiedades de Excel o con métodos poco robustos como VBA (Visual Basic Application)
- No se puede justificar y atribuir a alguien el cambio y/o modificación de un dato-valor
- La seguridad de la hoja se da con el bloqueo de la misma
- Se pueden copiar y pegar las hojas en otras rutas
- Uso de hojas multiusuario.
Integridad de datos
- No cuentan con Audit trail “pistas de auditoria” (elemento crítico para dar trazabilidad a cada movimiento realizado en un entorno GxP computarizado)
- No cuentan con la capacidad de respaldo y recuperación de la información
- Los datos no se asocian con bases de datos que hagan que estos perduren y, por tanto no pueden ser mantenidos; Atribuibles,legibles, contemporáneos y exactos (ALCOA).
Principio
Derivado de los puntos y controles sugeridos por las agencias regulatorias con el fin de garantizar la integridad de datos y con esto mantener el concepto de “ALCOA ++” partiendo de que los datos sean perdurables, se necesitan infraestructuras para garantizar la misma.
ALCOA ++, se atribuye a los siguientes términos:
- Atribuible, todas las actividades tales como crear, borrar o modificar son relacionadas a un individuo.
- Legible, la información debe ser clara y visible.
- Contemporáneo, la información debe ser registrada en el momento de hacer la actividad.
- Original, los datos deben provenir de registros originales o copias fieles a la original.
- Exactos, la exactitud debe ser controlada para eliminar errores y los cambios deben ser documentados.
- +Completo, todos los datos pertenecientes al registro GxP deberán ser retenidos.
- +Consistente, la línea del tiempo del metadato debe ser lógico.
- +Duradero, todos los registros deben estar en un medio duradero de manera legible durante su tiempo de retención.
- +Disponible, los datos deberán estar disponibles y accesibles en su totalidad.
Qué refiere una hoja de cálculo con integridad?
Las hojas de cálculo con integridad de datos, manifiestan un alto comportamiento en la trazabilidad y vida de un dato-valor, de tal manera que la perdurabilidad de este sea consistente y por tanto se pueda mantener: atribuible, legible, contemporáneo y exacto.
Suponga que está utilizando una hoja de cálculo y que necesita cambiar los valores obtenidos de al menos una prueba dentro de una fila. Tiene un pequeño problema: tiene que cambiar el valor de varias filas. Por ejemplo, el pH aparece en tres filas. Pero aparecerá un problema de verdad si olvida cambiar uno de los valores de las filas – el valor de pH asignado a esta fila se volverá ambigua, y por tanto sus datos perderán integridad.
Como hacer que una hoja de cálculo tenga Integridad de datos?
Las hojas de cálculo se deben evaluar y, con base en esto determinar el alcance GxP.

Derivado de la determinación, podemos considerar el diseño del sistema computarizado. Para esta transformación se necesitan procesos de programación mediante lenguajes robustos obtenidos por aplicaciones. Las hojas de cálculo más recientes con enfoque a entornos y control de datos GxP tiene estas capacidades.
CFR21 parte 11.10 «Controles»
Cuando hacemos referencias a hojas de cálculo, estas deben ser tratadas como un sistema computarizado. Por este hecho a esta herramienta le aplican los siguientes controles regulatorios y de cumplimiento:
- Debe permitir el proceso de validación para garantizar la precisión, la confiabilidad, el desempeño esperado consistente y la capacidad de discernir registros inválidos o alterados.
- Debe proporcionar la capacidad de generar copias precisas y completas de los registros en forma legible y en formato electrónico, para que la entidad reguladora pueda inspeccionarlos, revisarlos y copiarlos.
- Debe permitir la recuperación precisa , durante todo el período de retención de registros.
- Debe limitar el acceso del sistema a personas autorizadas.
- Debe usar pistas de auditoría seguras, generadas por computadora y con marca de tiempo, para registrar de manera independiente la fecha y la hora de las entradas y acciones del operador que crean, modifican o eliminan registros electrónicos. Los cambios de registro no deben ocultar la información previamente grabada.
- Debe permitir establecer el cumplimiento de políticas escritas, que responsabilizan a las personas de las acciones iniciadas bajo sus firmas electrónicas, con el fin de impedir la falsificación de registros y firmas.
- Debe proporcionar controles de autoridad, para garantizar que solo las personas autorizadas puedan usar el sistema, firmar electrónicamente un registro, acceder a la operación o al dispositivo de entrada o salida del sistema informático, alterar un registro o realizar operación en cuestión.
- Debe tener controles adecuados sobre la distribución, el acceso y el uso de la documentación para la garantizar su trazabilidad.
Por qué es importante la integridad de datos?
La importancia radica en disminuir el riesgo de que los datos críticos pueden ser falseados, borrados, divulgados sin autorización, modificados o negados por los emisores. Y con esto, no tener la posibilidad de trazar la historia y/o auditarla.
Lineamientos regulatorios a considerar
Todos los registros de calidad y técnico/científicos (incluyendo informes de análisis, certificados de análisis y hojas de trabajo analítico) deben ser legibles, rápidamente recuperables, almacenados y retenidos dentro de instalaciones que proporcionen un medio ambiente adecuado que prevenga modificaciones, daño o deterioro y/o pérdida.
Las condiciones bajo las cuales todos los registros originales son almacenados deben ser tales que aseguren su seguridad y confidencialidad, y el acceso a ellos debe estar restringido al personal autorizado. Se pueden emplear también almacenamiento y firmas electrónicos, pero con acceso restringido y en conformidad con los requisitos de los registros electrónicos.
Disposiciones asociadas a sistemas cerrados (incluye hojas de Excel)
- Limitar el acceso al sistema a personas autorizadas.
- Uso de verificaciones del sistema operativo.
- Uso de controles de autoridad.
- Determinación de que las persona que desarrollan , mantienen o usas sistemas tienen la educación , capacitación y experiencia para realizar las tareas asignadas.
- Establecimiento de políticas escritas que responsabilizan a las personas por las acciones iniciadas bajo sus firmas electrónicas.
- Controles apropiados sobre la documentación de los sistemas.
No olvides que en caso de ser necesario, puedes contactarnos para mejorar el proceso de laboratorio y administrativo de tu organización a través software 100% confiable y seguro. Recuerda que en deappharma somos expertos en la materia, por lo que procesos complejos no representa un reto para nosotros, debido a que tenemos en mente los aspectos relevantes de cumplimiento. ¡Conoce nuestras soluciones da clic aquí!
Referencias
- CFR 21 Parte 11.10 Closed systems
- FDA Data Integrity and Compliance With CGMP Guidance for Industry
- NOM 059 2015 Buenas prácticas de fabricación de medicamentos.
- WHO Technical Report Series, No. 957, 2010 44th Report – Annex 1 Buenas prácticas de la OMS para laboratorios de control de calidad de productos farmacéuticos
¡Contáctanos! y conoce de que manera te vamos a ayudar.
¡Únete a nuestra comunidad en redes!
29, Jun 2021
Revisión de estado validado de método analíticos.
Uno de los varios procesos de control analítico dentro de la industria, es el seguimiento o mantenimiento del estado validado y/o estado de control de métodos analíticos. No es una tarea fácil!! ya que para realizar una correcta revisión, el entorno en el que gira el proceso de mantenimiento validado radica en el acceso a documentos y en la obtención de datos que se asocian directamente a la esencia del método, como son: primera validación y parámetros de adecuabilidad históricos.
Concepto:
El concepto de ciclo de vida, vincula el desarrollo de productos y procesos, por lo que se debe adquirir conocimiento para abordar la revisión.
Validación métodos y calidad de los medicamentos.
La validación efectiva del método contribuye significativamente a asegurar la calidad de los medicamentos. Lo básico, es comprender el principio de garantía de calidad. Este principio describe que se debe producir un medicamento que sea apto para el uso previsto.
Este principio incorpora el entendimiento de que existen las siguientes condiciones:
a) La calidad, la seguridad y la eficacia están diseñadas o integradas en el producto.
b) La calidad no puede garantizarse adecuadamente simplemente mediante el producto en proceso y terminado, inspección o prueba.
Un programa de validación exitoso depende de la información y el conocimiento del método, así como, de la comprensión del proceso de desarrollo analítico. Este conocimiento es la base para establecer un enfoque para el control del analítico, que da como resultado certidumbre en la calidad y atributos evaluados. Los científicos analíticos deben:
- Comprender las fuentes de variación (se puede evaluar durante la validación o con la revisión de los datos de validación).
- Detectar la presencia y el grado de variación (análisis de datos obtenidos en la validación y en el histórico de datos de liberación, sin embargo, esto no mide directamente el método).
- Controlar la variación de una manera acorde con el riesgo, que representa para el método que afecta los datos de liberación del producto.
Descripción del proceso analítico en tres etapas:
Etapa 1 – Diseño del proceso analítico: el proceso analítico se define durante este etapa, basada en el conocimiento adquirido a través de actividades de desarrollo.
Etapa 2 – Validación del proceso analítico: durante esta etapa, el diseño analítico se evalúa para determinar si método es capaz de producir resultados reproducibles.
Etapa 3 – Verificación continua del proceso analítico: la garantía continua se obtiene durante la rutina analítica, de tal manera, que se pueda verificar que el proceso permanece en un estado de control.
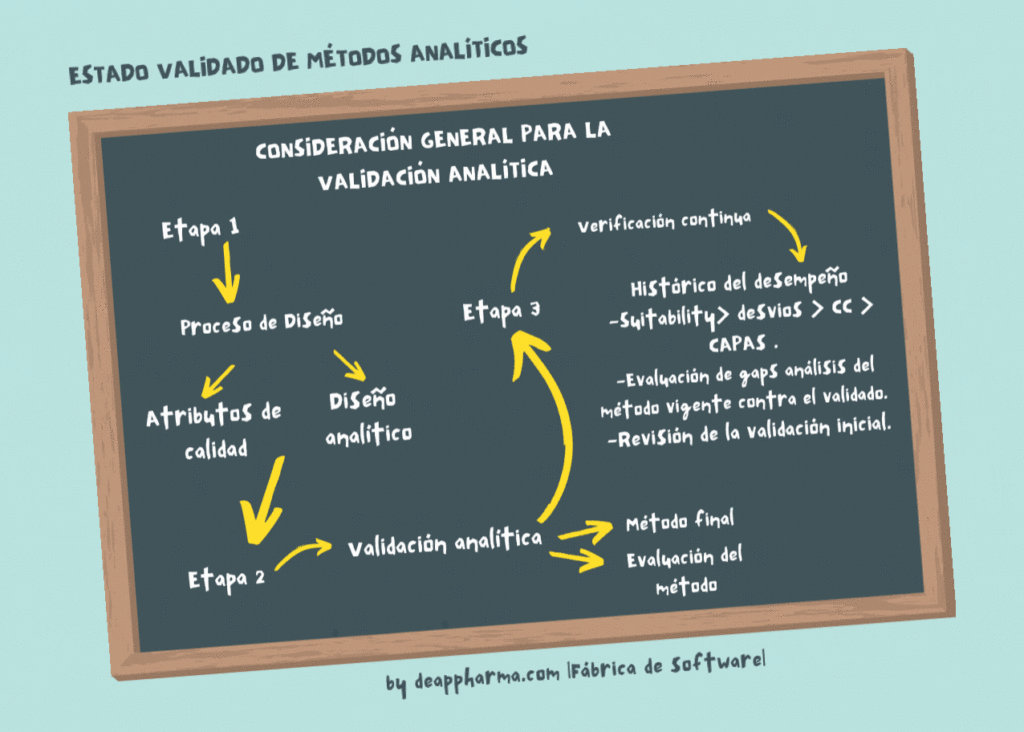
Los puntos relevantes de evaluación a la etapa 3, son:
- Requisitos de idoneidad históricos del sistema (platos teóricos, resolución, factor de coleo, factor de simetría, factor de capacidad y/o tiempos de retención de componentes).
- Parametrización y evaluación de las pruebas de desempeño acordes al método analítico.
- Revisión de los ajustes a las condiciones de operación indicadas en el procedimiento analítico vigente vs los descritos en la validación y/o transferencia inicial.
- Hallazgos o desviaciones, directamente relacionadas al desempeño del método analítico.
Si una evaluación basada en riesgos es utilizada como herramienta, se deben considerar otros factores. Los cuales pueden conducir a cambios en un procedimiento analítico y/o reemplazo con un nuevo método, se puede considerar los siguiente:
- Un ejercicio nuevo de transferencia;
- Una posible revalidación, sea parcial o total, o,un nuevo ejercicio de validación o,
- Un estudio de comparabilidad de métodos analíticos, es una opción factible.
En algunos casos, cambios en el fármaco o sustancias dentro del proceso de fabricación también deben ser evaluados como parte de la revisión de estado validado.
Recurso analítico «software».
Como se menciono anteriormente, la revisión de estado validado de un método analítico radica en que tan disponible y correcta fue la ejecución de este. Por lo que, la disposición de la información debe ser consistente. De otra manera será difícil concluir.
eADMxL el software de cumplimiento de deappharma.com , le ayudara con el control del proceso de estado validado mediante sus funciones especificas de «Revisión de estado validado» y «Monitoreo de vigencias».
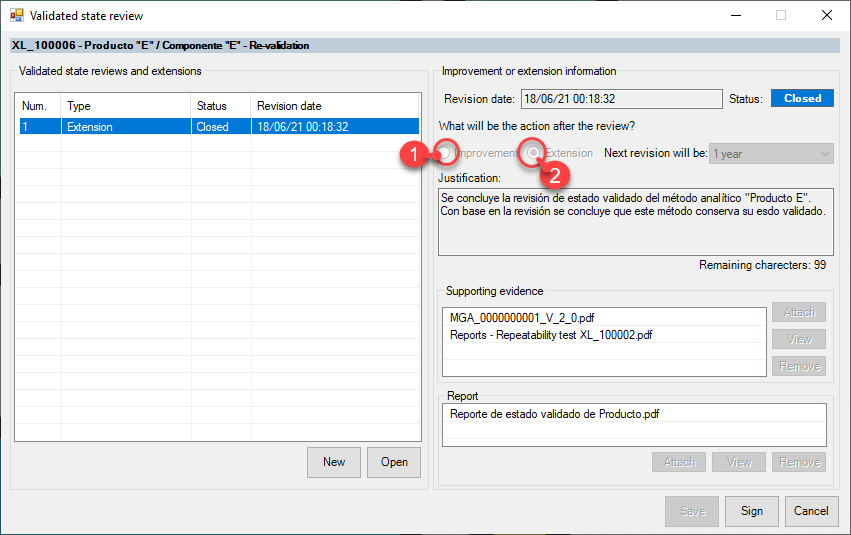
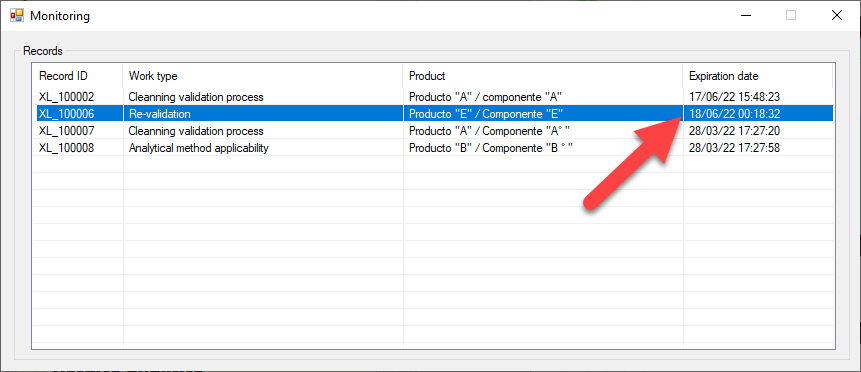
Algunas preguntas relevantes, que le ayudaran a definir el estado de control y/o mantenimiento, son:
- El método fue planteado y validado en concordancia con su categoría?
- La estadística paramétrica es integra?
- Los resultados de validación analítica son consistentes y claros?
- Si es un método compendial, los datos de pruebas de robustez tolerados están documentados?
- Los criterios de “Suitability” del sistema, se mantienen a través de los análisis?
- Las condiciones analíticas establecidas en el método final (preparación , condiciones de análisis, columna etc.) son las mismas que están descritas en el reporte de validación del método?
- Si hay cambios al método, se registran y se alinean a un control de cambio y se emite un CaPa al método y con esto evaluar el impacto a la validación?
- Qué tan recurrentes son las desviaciones o justificaciones asociadas al método analítico?
- Si la validación analítica sigue vigente, puedo hacer un revisión de estado validado antes de su fin expiración si surgen problemas?
No olvides que en caso de ser necesario, puedes contactarnos para mejorar el proceso de laboratorio y administrativo de tu organización a través software 100% confiable y seguro. Recuerda que en deappharma somos expertos en la materia, por lo que procesos complejos no representa un reto para nosotros, debido a que tenemos en mente los aspectos relevantes de cumplimiento.
Referencias.
- Analytical Procedures and Methods Validation for Drugs and Biologics Guidance for Industry
- Pharmaceutical quality system ICH Q10.
- Guidance for Industry Process Validation: General Principles and Practices.
¡Contáctanos! y conoce de que manera te vamos a ayudar.
¡Únete a nuestra comunidad en redes!